
Title: Radio-Activity
Author: Ernest Rutherford
Release date: March 4, 2021 [eBook #64693]
Most recently updated: September 17, 2023
Language: English
Credits: Richard Tonsing, David King, and the Online Distributed Proofreading Team at http://www.pgdp.net. (This file was produced from images generously made available by The Internet Archive.)
In this work, I have endeavoured to give a complete and connected account, from a physical standpoint, of the properties possessed by the naturally radio-active bodies. Although the subject is comparatively a new one, our knowledge of the properties of the radio-active substances has advanced with great rapidity, and there is now a very large amount of information on the subject scattered throughout the various scientific journals.
The phenomena exhibited by the radio-active bodies are extremely complicated, and some form of theory is essential in order to connect in an intelligible manner the mass of experimental facts that have now been accumulated. I have found the theory that the atoms of the radio-active bodies are undergoing spontaneous disintegration extremely serviceable, not only in correlating the known phenomena, but also in suggesting new lines of research.
The interpretation of the results has, to a large extent, been based on the disintegration theory, and the logical deductions to be drawn from the application of the theory to radio-active phenomena have also been considered.
The rapid advance of our knowledge of radio-activity has been dependent on the information already gained by research into the electric properties of gases. The action possessed by the radiations from radio-active bodies of producing charged carriers or ions in the gas, has formed the basis of an accurate quantitative method of examination of the properties of the radiations and of viiiradio-active processes, and also allows us to determine with considerable certainty the order of magnitude of the different quantities involved.
For these reasons, it has been thought advisable to give a brief account of the electric properties of gases, to the extent that is necessary for the interpretation of the results of measurements in radio-activity by the electric method. The chapter on the ionization theory of gases was written before the publication of J. J. Thomson’s recent book on “Conduction of Electricity through Gases,” in which the whole subject is treated in a complete and connected manner.
A short chapter has been added, in which an account is given of the methods of measurement which, in the experience of the writer and others, are most suitable for accurate work in radio-activity. It is hoped that such an account may be of some service to those who may wish to obtain a practical acquaintance with the methods employed in radio-active measurements.
My thanks are due to Mr W. C. Dampier Whetham, F.R.S., one of the editors of the Cambridge Physical Series, for many valuable suggestions, and for the great care and trouble he has taken in revising the proof sheets. I am also much indebted to my wife and Miss H. Brooks for their kind assistance in correcting the proofs, and to Mr R. K. McClung for revising the index.
I feel that some apology is due to my readers for bringing out at such an early date a new edition which includes so much new material, and in which the rearrangement is so extensive as to constitute almost a new work. Though only a year has passed since the book first made its appearance, the researches that have been carried out in that time have been too numerous and of too important a character to permit the publishing of a mere reprint, unless the author were to relinquish his purpose of presenting the subject as it stands at the present moment.
The three new chapters which have been added possibly constitute the most important change in the work. These chapters include a detailed account of the theory of successive changes and of its application to the analysis of the series of transformations which occur in radium, thorium, and actinium.
The disintegration theory, which was put forward in the first edition as an explanation of radio-active phenomena, has in these later researches proved to be a most powerful and valuable method of analysing the connection between the series of substances which arise from the transformation of the radio-elements. It has disclosed the origin of radium, of polonium and radio-tellurium, and of radio-lead, and now binds together in one coherent whole the large mass of apparently heterogeneous experimental facts in radio-activity which have been accumulating since 1896. The theory has received a remarkable measure of verification in the past year, and, in many cases, has offered a quantitative as well xas a qualitative explanation of the connection between the various properties exhibited by the radio-active bodies. In the light of this evidence, radio-activity may claim to have assumed the position of an independent subject, though one with close affinities to physics on the one hand and to chemistry on the other.
The present edition includes a large amount of new material relating to the nature and properties of the radiations and the emanations. In the limits of this book, it would have been found impossible, even had it been thought desirable, to include more than a brief sketch of the physiological effects of the rays. The literature on this subject is already large, and is increasing rapidly. For reasons of space, I have not been able to refer more than briefly to the mass of papers that have appeared dealing with the examination of various spring and well waters, sediments, and soils, for the presence of radio-active matter.
In order to make the book more self-contained, a short account has been given in Chapter II of the magnetic field produced by an ion in motion, of the action of an external magnetic and electric field upon it, and of the determination of the velocity and mass of the particles constituting the cathode stream.
Two appendices have been added, one giving an account of some work upon the α rays which was completed too late for inclusion in the subject matter of the book, and the other containing a brief summary of what is known in regard to the chemical constitution of the various radio-active minerals, the localities in which they are found, and their probable geologic age. For the preparation of the latter, I am indebted to my friend Dr Boltwood of New Haven, who, in the course of his researches, has had occasion to analyse most of these minerals in order to determine their content of uranium and radium. I hope that this account of radio-active minerals will prove of value to those who are endeavouring to elucidate the connection between the various radio-active substances and the inactive products which arise from their transformation.
xiFor the convenience of those who have read the first edition, a list of the sections and chapters which contain the most important additions and alterations is added below the table of contents.
The writing of a complete account of a subject like radio-activity, in which so much new work is constantly appearing, has been a matter of no little difficulty. Among other things it has involved a continuous revision of the work while the volume was passing through the press.
I wish to express my thanks to my colleague Professor Harkness for the care and trouble he has taken in revising the proofs and for many useful suggestions; also to Mr R. K. McClung for his assistance in correcting some of the proofs and in preparing the index.
These corrections have been applied to the text in the book.
II. Ionization Theory of Gases 31
III. Methods of Measurement 82
IV. Nature of the Radiations 108
V. Properties of the Radiations 201
VI. Continuous Production of Radio-active Matter 218
VII. Radio-active Emanations 238
VIII. Excited Radio-activity 295
IX. Theory of Successive Changes 325
X. Transformation Products of Uranium, Thorium and Actinium 346
XI. Transformation Products of Radium 371
XII. Rate of Emission of Energy 418
XIII. Radio-active Processes 437
XIV. Radio-activity of the Atmosphere and of Ordinary Materials 501
Appendix A. Properties of the α Rays 543
Appendix B. Radio-active Minerals 554
Plate (Fig. 46A: Spectrum of Radium Bromide) to face p. 206
For the convenience of the reader, the sections and chapters which contain mostly new matter, or have been either partly or wholly rewritten, are appended below.
xivABBREVIATIONS OF REFERENCES TO SOME OF THE JOURNALS.
Ber. d. deutsch. Chem. Ges. Berichte der deutschen chemischen Gesellschaft. Berlin.
C. R. Comptes Rendus des Séances de l’Académie des Sciences. Paris.
Chem. News. Chemical News. London.
Drude’s Annal. Annalen der Physik. Leipzig.
Phil. Mag. Philosophical Magazine and Journal of Science. London.
Phil. Trans. Philosophical Transactions of the Royal Society of London.
Phys. Rev. Physical Review. New York.
Phys. Zeit. Physikalische Zeitschrift.
Proc. Camb. Phil. Soc. Proceedings of the Cambridge Philosophical Society. Cambridge.
Proc. Roy. Soc. Proceedings of the Royal Society of London.
Thèses-Paris. Thèses présentées à la Faculté des Sciences de l’Université de Paris.
Wied. Annal. Annalen der Physik. Leipzig.
1. Introduction. The close of the old and the beginning of the new century have been marked by a very rapid increase of our knowledge of that most important but comparatively little known subject—the connection between electricity and matter. No study has been more fruitful in surprises to the investigator, both from the remarkable nature of the phenomena exhibited and from the laws controlling them. The more the subject is examined, the more complex must we suppose the constitution of matter in order to explain the remarkable effects observed. While the experimental results have led to the view that the constitution of the atom itself is very complex, at the same time they have confirmed the old theory of the discontinuous or atomic structure of matter. The study of the radio-active substances and of the discharge of electricity through gases has supplied very strong experimental evidence in support of the fundamental ideas of the existing atomic theory. It has also indicated that the atom itself is not the smallest unit of matter, but is a complicated structure made up of a number of smaller bodies.
A great impetus to the study of this subject was initially given by the experiments of Lenard on the cathode rays, and by Röntgen’s discovery of the X rays. An examination of the conductivity imparted to a gas by the X rays led to a clear view of the mechanism of the transport of electricity through gases by means of charged ions. This ionization theory of gases has been shown to afford a satisfactory explanation not only of the passage of electricity through flames and vapours, but also of the 2complicated phenomena observed when a discharge of electricity passes through a vacuum tube. At the same time, a further study of the cathode rays showed that they consisted of a stream of material particles, projected with great velocity, and possessing an apparent mass small compared with that of the hydrogen atom. The connection between the cathode and Röntgen rays and the nature of the latter were also elucidated. Much of this admirable experimental work on the nature of the electric discharge has been done by Professor J. J. Thomson and his students in the Cavendish Laboratory, Cambridge.
An examination of natural substances, in order to see if they gave out dark radiations similar to X rays, led to the discovery of the radio-active bodies which possess the property of spontaneously emitting radiations, invisible to the eye, but readily detected by their action on photographic plates and their power of discharging electrified bodies. A detailed study of the radio-active bodies has revealed many new and surprising phenomena which have thrown much light, not only on the nature of the radiations themselves, but also on the processes occurring in those substances. Notwithstanding the complex nature of the phenomena, the knowledge of the subject has advanced with great rapidity, and a large amount of experimental data has now been accumulated.
In order to explain the phenomena of radio-activity, Rutherford and Soddy have advanced a theory which regards the atoms of the radio-active elements as suffering spontaneous disintegration, and giving rise to a series of radio-active substances which differ in chemical properties from the parent elements. The radiations accompany the breaking-up of the atoms, and afford a comparative measure of the rate at which the disintegration takes place. This theory is found to account in a satisfactory way for all the known facts of radio-activity, and welds a mass of disconnected facts into one homogeneous whole. On this view, the continuous emission of energy from the active bodies is derived from the internal energy inherent in the atom, and does not in any way contradict the law of the conservation of energy. At the same time, however, it indicates that an enormous store of latent energy is resident in the radio-atoms themselves. This store of energy has not been observed previously, on account of the impossibility of breaking up 3into simpler forms the atoms of the elements by the action of the chemical or physical forces at our command.
On this theory we are witnessing in the radio-active bodies a veritable transformation of matter. This process of disintegration was investigated, not by direct chemical methods, but by means of the property possessed by the radio-active bodies of giving out specific types of radiation. Except in the case of a very active element like radium, the process of disintegration takes place so slowly, that hundreds if not thousands of years would be required before the amount transformed would come within the range of detection of the balance or the spectroscope. In radium, however, the process of disintegration takes place at such a rate that it should be possible within a limited space of time to obtain definite chemical evidence on this question. The recent discovery that helium can be obtained from radium adds strong confirmation to the theory; for helium was indicated as a probable disintegration product of the radio-active elements before this experimental evidence was forthcoming. Several products of the transformation of the radio-active bodies have already been examined, and the further study of these substances promises to open up new and important fields of chemical enquiry.
In this book the experimental facts of radio-activity and the connection between them are interpreted on the disintegration theory. Many of the phenomena observed can be investigated in a quantitative manner, and prominence has been given to work of this character, for the agreement of any theory with the facts, which it attempts to explain, must ultimately depend upon the results of accurate measurement.
The value of any working theory depends upon the number of experimental facts it serves to correlate, and upon its power of suggesting new lines of work. In these respects the disintegration theory, whether or not it may ultimately be proved to be correct, has already been justified by its results.
2. Radio-active Substances. The term “radio-active” is now generally applied to a class of substances, such as uranium, thorium, radium, and their compounds, which possess the property of spontaneously emitting radiations capable of passing through 4plates of metal and other substances opaque to ordinary light. The characteristic property of these radiations, besides their penetrating power, is their action on a photographic plate and their power of discharging electrified bodies. In addition, a strongly radio-active body like radium is able to cause marked phosphorescence and fluorescence on some substances placed near it. In the above respects the radiations possess properties analogous to Röntgen rays, but it will be shown that, for the major part of the radiations emitted, the resemblance is only superficial.
The most remarkable property of the radio-active bodies is their power of radiating energy spontaneously and continuously at a constant rate, without, as far as is known, the action upon them of any external exciting cause. The phenomena at first sight appear to be in direct contradiction to the law of conservation of energy, since no obvious change with time occurs in the radiating material. The phenomena appear still more remarkable when it is considered that the radio-active bodies must have been steadily radiating energy since the time of their formation in the earth’s crust.
Immediately after Röntgen’s discovery of the production of X rays, several physicists were led to examine if any natural bodies possessed the property of giving out radiations which could penetrate metals and other substances opaque to light. As the production of X rays seemed to be connected in some way with cathode rays, which cause strong fluorescent and phosphorescent effects on various bodies, the substances first examined were those that were phosphorescent when exposed to light. The first observation in this direction was made by Niewenglowski[1], who found that sulphide of calcium exposed to the sun’s rays gave out some rays which were able to pass through black paper. A little later a similar result was recorded by H. Becquerel[2] for a special calcium sulphide preparation, and by Troost[3] for a specimen of hexagonal blend. These results were confirmed and extended in a later paper by Arnold[4]. No satisfactory explanations of these 5somewhat doubtful results have yet been given, except on the view that the black paper was transparent to some of the light waves. At the same time Le Bon[5] showed that, by the action of sunlight on certain bodies, a radiation was given out, invisible to the eye, but active with regard to a photographic plate. These results have been the subject of much discussion; but there seems to be little doubt that the effects are due to short ultra-violet light waves, capable of passing through certain substances opaque to ordinary light. These effects, while interesting in themselves, are quite distinct in character from those shown by the radio-active bodies which will now be considered.
3. Uranium. The first important discovery in the subject of radio-activity was made in February, 1896, by M. Henri Becquerel[6], who found that a uranium salt, the double sulphate of uranium and potassium, emitted some rays which gave an impression on a photographic plate enveloped in black paper. These rays were also able to pass through thin plates of metals and other substances opaque to light. The impressions on the plate could not have been due to vapours given off by the substances, since the same effect was produced whether the salt was placed directly on the black paper or on a thin plate of glass lying upon it.
Becquerel found later that all the compounds of uranium as well as the metal itself possessed the same property, and, although the amount of action varied slightly for the different compounds, the effects in all cases were comparable. It was at first natural to suppose that the emission of these rays was in some way connected with the power of phosphorescence, but later observations showed that there was no connection whatever between them. The uranic salts are phosphorescent, while the uranous salts are not. The uranic salts, when exposed to ultra-violet light in the phosphoroscope, give a phosphorescent light lasting about ·01 seconds. When the salts are dissolved in water, the duration is still less. The amount of action on the photographic plate does not depend on the particular compound of uranium employed, but only on the amount of uranium present in the compound. The non-phosphorescent are 6equally active with the phosphorescent compounds. The amount of radiation given out is unaltered if the active body be kept continuously in darkness. The rays are given out by solutions, and by crystals which have been deposited from solutions in the dark and never exposed to light. This shows that the radiation cannot be due in any way to the gradual emission of energy stored up in the crystal in consequence of exposure to a source of light.
4. The power of giving out penetrating rays thus seems to be a specific property of the element uranium, since it is exhibited by the metal as well as by all its compounds. These radiations from uranium are persistent, and, as far as observations have yet gone, are unchanged, either in intensity or character, with lapse of time. Observations to test the constancy of the radiations for long periods of time have been made by Becquerel. Samples of uranic and uranous salts have been kept in a double box of thick lead, and the whole has been preserved from exposure to light. By a simple arrangement, a photographic plate can be introduced in a definite position above the uranium salts, which are covered with a layer of black paper. The plate is exposed at intervals for 48 hours, and the impression on the plate compared. No perceptible weakening of the radiation has been observed over a period of four years. Mme Curie[7] has made determinations of the activity of uranium over a space of five years by an electric method described later, but found no appreciable variation during that period.
Since the uranium is thus continuously radiating energy from itself, without any known source of excitation, the question arises whether any known agent is able to affect the rate of its emission. No alteration was observed when the body was exposed to ultra-violet light or to ultra-red light or to X rays. Becquerel states that the double sulphate of uranium and potassium showed a slight increase of action when exposed to the arc light and to sparks, but he considers that the feeble effect observed was another action superimposed on the constant radiation from uranium. The intensity of the uranium radiation is not affected by a variation of temperature between 200° C. and the temperature of liquid air. This question is discussed in more detail later.
75. In addition to these actions on a photographic plate, Becquerel showed that uranium rays, like Röntgen rays, possess the important property of discharging both positively and negatively electrified bodies. These results were confirmed and extended by Lord Kelvin, Smolan and Beattie[8]. The writer made a detailed comparison[9] of the nature of the discharge produced by uranium with that produced by Röntgen rays, and showed that the discharging property of uranium is due to the production of charged ions by the radiation throughout the volume of the gas. The property has been made the basis of a qualitative and quantitative examination of the radiations from all radio-active bodies, and is discussed in detail in chapter II.
The radiations from uranium are thus analogous, as regards their photographic and electrical actions, to Röntgen rays, but, compared with the rays from an ordinary X ray tube, these actions are extremely feeble. While with Röntgen rays a strong impression is produced on a photographic plate in a few minutes or even seconds, several days’ exposure to the uranium rays is required to produce a well-marked action, even though the uranium compound, enveloped in black paper, is placed close to the plate. The discharging action, while very easily measurable by suitable methods, is also small compared with that produced by X rays from an ordinary tube.
6. The rays from uranium show no evidence of direct reflection, refraction, or polarization[10]. While there is no direct reflection of the rays, there is apparently a diffuse reflection produced where the rays strike a solid obstacle. This is due in reality to a secondary radiation set up when the primary rays impinge upon matter. The presence of this secondary radiation at first gave rise to the erroneous view that the rays could be reflected and refracted like ordinary light. The absence of reflection, refraction, or polarization in the penetrating rays from uranium necessarily follows in the light of our present knowledge of the rays. It is now known that the uranium rays, mainly responsible for the photographic action, are deviable by a magnetic field, and 8are similar in all respects to cathode rays, i.e. the rays are composed of small particles projected at great velocities. The absence of the ordinary properties of transverse light waves is thus to be expected.
7. The rays from uranium are complex in character, and, in addition to the penetrating deviable rays, there is also given off a radiation very readily absorbed by passing through thin layers of metal foil, or by traversing a few centimetres of air. The photographic action due to these rays is very feeble in comparison with that of the penetrating rays, although the discharge of electrified bodies is mainly caused by them. Besides these two types of rays, some rays are emitted which are of an extremely penetrating character and are non-deviable by a magnetic field. These rays are difficult to detect photographically, but can readily be examined by the electric method.
8. The question naturally arose whether the property of spontaneously giving out penetrating radiations was confined to uranium and its compounds, or whether it was exhibited to any appreciable extent by other substances.
By the electrical method, with an electrometer of ordinary sensitiveness, any body which possesses an activity of the order of ¹⁄₁₀₀ of that of uranium can be detected. With an electroscope of special construction, such as has been designed by C. T. R. Wilson for his experiments on the natural ionization of air, a substance of activity ¹⁄₁₀₀₀₀ and probably ¹⁄₁₀₀₀₀₀ of that of uranium can be detected.
If an active body like uranium be mixed with an inactive body, the resulting activity in the mixture is generally considerably less than that due to the active substance alone. This is due to the absorption of the radiation by the inactive matter present. The amount of decrease largely depends on the thickness of the layer from which the activity is determined.
Mme Curie made a detailed examination by the electrical method of the great majority of known substances, including the very rare elements, to see if they possessed any activity. In cases where it was possible, several compounds of the elements were examined. With the exception of thorium and phosphorus, none 9of the other substances possessed an activity even of the order of ¹⁄₁₀₀ of uranium.
The ionization of the gas by phosphorus does not, however, seem to be due to a penetrating radiation like that found in the case of uranium, but rather to a chemical action taking place at its surface. The compounds of phosphorus do not show any activity, and in this respect differ from uranium and the other active bodies.
Le Bon[11] has also observed that quinine sulphate, if heated and then allowed to cool, possesses for a short time the property of discharging both positively and negatively electrified bodies. It is necessary, however, to draw a sharp line of distinction between phenomena of this kind and those exhibited by the naturally radio-active bodies. While both, under special conditions, possess the property of ionizing the gas, the laws controlling the phenomena are quite distinct in the two cases. For example, only one compound of quinine shows the property, and that compound only when it has been subjected to a preliminary heating. The action of phosphorus depends on the nature of the gas, and varies with temperature. On the other hand, the activity of the naturally radio-active bodies is spontaneous and permanent. It is exhibited by all compounds, and is not, as far as is yet known, altered by change in the chemical or physical conditions.
9. The discharging and photographic action alone cannot be taken as a criterion as to whether a substance is radio-active or not. It is necessary in addition to examine the radiations, and to test whether the actions take place through appreciable thicknesses of all kinds of matter opaque to ordinary light. For example, a body giving out short waves of ultra-violet light can be made to behave in many respects like a radio-active body. As Lenard[12] has shown, short waves of ultra-violet light will ionize the gas in their path, and will be absorbed rapidly in the gas. They will produce strong photographic action, and may pass through some substances opaque to ordinary light. The similarity to a radio-active body is thus fairly complete as regards these properties. On the other 10hand, the emission of these light waves, unlike that of the radiations from an active body, will depend largely on the molecular state of the compound, or on temperature and other physical conditions. But the great point of distinction lies in the nature of the radiations from the bodies in question. In one case the radiations behave as transverse waves, obeying the usual laws of light waves, while in the case of a naturally active body, they consist for the most part of a continuous flight of material particles projected from the substance with great velocity. Before any substance can be called “radio-active” in the sense in which the term is used to describe the properties of the natural radio-active elements, it is thus necessary to make a close examination of its radiation; for it is unadvisable to extend the use of the term “radio-active” to substances which do not possess the characteristic radiating properties of the radio-active elements which we have described, and the active products which can be obtained from them. Some of the pseudo-active bodies will however be considered later in chapter IX.
10. Thorium. In the course of an examination of a large number of substances, Schmidt[13] found that thorium, its compounds, and the minerals containing thorium, possessed properties similar to those of uranium. The same discovery was made independently by Mme Curie[14]. The rays from thorium compounds, like those from uranium, possess the properties of discharging electrified bodies and acting on a photographic plate. Under the same conditions the discharging action of the rays is about equal in amount to that of uranium, but the photographic effect is distinctly weaker.
The radiations from thorium are more complicated than those from uranium. It was early observed by several experimenters that the radiation from thorium compounds, especially the oxide, when tested by the electrified method, was very variable and uncertain. A detailed investigation of the radiations from thorium under various conditions was made by Owens[15]. He showed that thorium oxide, especially in thick layers, was able to produce 11conductivity in the gas when covered with a large thickness of paper, and that the amount of this conductivity could be greatly varied by blowing a current of air over the gas. In the course of an examination[16] of this action of the air current, the writer showed that thorium compounds gave out a material emanation made up of very small particles themselves radio-active. The emanation behaves like a radio-active gas; it diffuses rapidly through porous substances like paper, and is carried away by a current of air. The evidence of the existence of the emanation and its properties, is considered in detail later in chapter VIII. In addition to giving out an emanation, thorium behaves like uranium in emitting three types of radiation, each of which is similar in properties to the corresponding radiation from uranium.
11. Radio-active minerals. Mme Curie has examined the radio-activity of a large number of minerals containing uranium and thorium. The electrical method was used, and the current measured between two parallel plates 8 cms. in diameter and 3 cms. apart, when one plate was covered with a uniform layer of the active matter. The following numbers give the order of the saturation current obtained in amperes.
| Pitchblende from Johanngeorgenstadt | 8·3 × 10-11 |
| „ Joachimsthal | 7·0 „ |
| „ Pzibran | 6·5 „ |
| „ Cornwall | 1·6 „ |
| Cleveite | 1·4 „ |
| Chalcolite | 5·2 „ |
| Autunite | 2·7 „ |
| Thorite | from 0·3 to 1·4 „ |
| Orangite | 2·0 „ |
| Monazite | 0·5 „ |
| Xenotine | 0·03 „ |
| Aeschynite | 0·7 „ |
| Fergusonite | 0·4 „ |
| Samarskite | 1·1 „ |
| Niobite | 0·3 „ |
| Carnotite | 6·2 „ |
Some activity is to be expected in these minerals, since they all contain either thorium or uranium or a mixture of both. An 12examination of the action of the uranium compounds with the same apparatus and under the same conditions led to the following results:
| Uranium (containing a little carbon) | 2·3 × 10-11 amperes |
| Black oxide of uranium | 2·6 „ |
| Green „ „ | 1·8 „ |
| Acid uranic hydrate | 0·6 „ |
| Uranate of sodium | 1·2 „ |
| Uranate of potassium | 1·2 „ |
| Uranate of ammonia | 1·3 „ |
| Uranous sulphate | 0·7 „ |
| Sulphate of uranium and potassium | 0·7 „ |
| Acetate | 0·7 „ |
| Phosphate of copper and uranium | 0·9 „ |
| Oxysulphide of uranium | 1·2 „ |
The interesting point in connection with these results is that some specimens of pitchblende have four times the activity of the metal uranium; chalcolite, the crystallized phosphate of copper and uranium, is twice as active as uranium; and autunite, a phosphate of calcium and uranium, is as active as uranium. From the previous considerations, none of the substances should have shown as much activity as uranium or thorium. In order to be sure that the large activity was not due to the particular chemical combination, Mme Curie prepared chalcolite artificially, starting with pure products. This artificial chalcolite had the activity to be expected from its composition, viz. about 0·4 of the activity of the uranium. The natural mineral chalcolite is thus five times as active as the artificial mineral.
It thus seemed probable that the large activity of some of these minerals, compared with uranium and thorium, was due to the presence of small quantities of some very active substance, which was different from the known bodies thorium and uranium.
This supposition was completely verified by the work of M. and Mme Curie, who were able to separate from pitchblende by purely chemical methods two active bodies, one of which in the pure state is over a million times more active than the metal uranium.
This important discovery was due entirely to the property of radio-activity possessed by the new bodies. The only guide 13in their separation was the activity of the products obtained. In this respect the discovery of these bodies is quite analogous to the discovery of rare elements by the methods of spectrum analysis. The method employed in the separation consisted in examining the relative activity of the products after chemical treatment. In this way it was seen whether the radio-activity was confined to one or another of the products, or divided between both, and in what ratio such division occurred.
The activity of the specimens thus served as a basis of rough qualitative and quantitative analysis, analogous in some respects to the indication of the spectroscope. To obtain comparative data it was necessary to test all the products in the dry state. The chief difficulty lay in the fact that pitchblende is a very complex mineral, and contains in varying quantities nearly all the known metals.
12. Radium. The analysis of pitchblende by chemical methods, using the procedure sketched above, led to the discovery of two very active bodies, polonium and radium. The name polonium was given to the first substance discovered by Mme Curie in honour of the country of her birth. The name radium was a very happy inspiration of the discoverers, for this substance in the pure state possesses the property of radio-activity to an astonishing degree.
Radium is extracted from pitchblende by the process used to separate barium, to which radium is very closely allied in chemical properties[17]. After the removal of other substances, the radium remains behind mixed with barium. It can, however, be partially separated from the latter by the difference in solubility of the chlorides in water, alcohol, or hydrochloric acid. The chloride of radium is less soluble than that of barium, and can be separated from it by the method of fractional crystallization. After a large number of precipitations, the radium can be freed almost completely from the barium.
Both polonium and radium exist in infinitesimal quantities in pitchblende. In order to obtain a few decigrammes of very active radium, it is necessary to use several tons of pitchblende, or the 14residues obtained from the treatment of uranium minerals. It is thus obvious that the expense and labour involved in preparation of a minute quantity of radium are very great.
M. and Mme Curie were indebted for their first working material to the Austrian government, who generously presented them with a ton of the treated residue of uranium materials from the State manufactory of Joachimsthal in Bohemia. With the assistance of the Academy of Science and other societies in France, funds were given to carry out the laborious work of separation. Later the Curies were presented with a ton of residues from the treatment of pitchblende by the Société Centrale de Produits Chimiques of Paris. The generous assistance afforded in this important work is a welcome sign of the active interest taken in these countries in the furthering of purely scientific research.
The rough concentration and separation of the residues was performed in the chemical works, and there followed a large amount of labour in purification and concentration. In this manner, the Curies were able to obtain a small quantity of radium which was enormously active compared with uranium. No definite results have yet been given on the activity of pure radium, but the Curies estimate that it is about one million times that of uranium, and may possibly be still higher. The difficulty of making a numerical estimate for such an intensely active body is very great. In the electric method, the activities are compared by noting the relative strength of the maximum or saturation current between two parallel plates, on one of which the active substance is spread. On account of the intense ionization of the gas between the plates, it is not possible to reach the saturation current unless very high voltages are applied. Approximate comparisons can be made by the use of metal screens to cut down the intensity of the radiations, if the proportion of the radiation transmitted by such a screen has been determined by direct experiment on impure material of easily measurable activity. The value of the activity of radium compared with that of uranium will however vary to some extent according to which of the three types of rays is taken as a basis of comparison.
It is thus difficult to control the final stages of the purification of radium by measurements of its activity alone. Moreover the activity of radium immediately after its preparation is only about 15one-fourth of its final value; it gradually rises to a maximum after the radium salt has been kept in the dry state for about a month. For control experiments in purification, it is advisable to measure the initial rather than the final activity.
Mme Curie has utilized the coloration of the crystals of radiferous barium as a means of controlling the final process of purification. The crystals of salts of radium and barium deposited from acid solutions are indistinguishable by the eye. The crystals of radiferous barium are at first colourless, but, in the course of a few hours, become yellow, passing to orange and sometimes to a beautiful rose colour. The rapidity of this coloration depends on the amount of barium present. Pure radium crystals do not colour, or at any rate not as rapidly as those containing barium. The coloration is a maximum for a definite proportion of radium, and this fact can be utilized as a means of testing the amount of barium present. When the crystals are dissolved in water the coloration disappears.
Giesel[18] has observed that pure radium bromide gives a beautiful carmine colour to the Bunsen flame. If barium be present in any quantity, only the green colour due to barium is observed, and a spectroscopic examination shows only the barium lines. This carmine coloration of the Bunsen flame is a good indication of the purity of the radium.
Since the preliminary announcement of the discovery of radium, Giesel[19] has devoted a great deal of attention to the separation of radium, polonium and other active bodies from pitchblende. He was indebted for his working material to the firm of P. de Haen, of Hanover, who presented him with a ton of pitchblende residues. Using the method of fractional crystallization of the bromide instead of the chloride, he has been able to prepare considerable quantities of pure radium. By this means the labour of final purification of radium has been much reduced. He states that six or eight crystallizations with the bromide are sufficient to free the radium almost completely from the barium.
13. Spectrum of radium. It was of great importance to settle as soon as possible whether radium was in reality modified 16barium or a new element with a definite spectrum. For this purpose the Curies prepared some specimens of radium chloride, and submitted them for examination of their spectrum to Demarçay, an authority on that subject. The first specimen of radium chloride examined by Demarçay[20] was not very active, but showed, besides the lines due to barium, a very strong new line in the ultra-violet. In another sample of greater activity, the line was still stronger and others also appeared, while the intensity of the new lines was comparable with those present due to barium. With a still more active specimen which was probably nearly pure, only three strong lines of barium appeared, while the new spectrum was very bright. The following table shows the wave-length of the new lines observed for radium. The wave lengths are expressed in Ångström units and the intensity of each ray is denoted by a number, the ray of maximum intensity being 16.
| Wave length | Intensity | Wave length | Intensity |
|---|---|---|---|
| 4826·3 | 10 | 4600·3 | 3 |
| 4726·9 | 5 | 4533·5 | 9 |
| 4699·6 | 3 | 4436·1 | 6 |
| 4692·1 | 7 | 4340·6 | 12 |
| 4683·0 | 14 | 3814·7 | 16 |
| 4641·9 | 4 | 3649·6 | 12 |
The lines are all sharply defined, and three or four of them have an intensity comparable with any known lines of other substances. There are also present in the spectrum two strong nebulous bands. In the visible part of the spectrum, which has not been photographed, the only noticeable ray has a wave length 5665, which is, however, very feeble compared with that of wave length 4826·3. The general aspect of the spectrum is similar to that of the alkaline earths; it is known that these metals have strong lines accompanied by nebulous bands.
The principal line due to radium can be distinguished in impure radium of activity 50 times that of uranium. By the electrical method it is easy to distinguish the presence of radium in a body which has an activity only ¹⁄₁₀₀ of uranium. With a more sensitive electrometer ¹⁄₁₀₀₀₀ of the activity of uranium 17could be observed. For the detection of radium, the examination of the radio-activity is thus a process nearly a million times more sensitive than spectrum analysis.
Later observations on the spectrum of radium have been made by Runge[21], Exner and Haschek[22], with specimens of radium prepared by Giesel. Crookes[23] has photographed the spectrum of radium in the ultra-violet, while Runge and Precht[24], using a highly purified sample of radium, observed a number of new lines in the spark spectrum. It has been mentioned already that the bromide of radium gives a characteristic pure carmine-red coloration to the Bunsen flame. The flame spectrum shows two broad bright bands in the orange-red, not observed in Demarçay’s spectrum. In addition there is a line in the blue-green and two feeble lines in the violet.
14. Atomic weight of radium. Mme Curie has made successive determinations of the atomic weight of the new element with specimens of steadily increasing purity. In the first observation the radium was largely mixed with barium, and the atomic weight obtained was the same as that of barium, 137·5. In successive observations with specimens of increasing purity the atomic weights of the mixture were 146 and 175. The final value obtained recently was 225, which may be taken as the atomic weight of radium on the assumption that it is divalent.
In these experiments about 0·1 gram of pure radium chloride was obtained by successive fractionations. The difficulty involved in preparing a quantity of pure radium chloride large enough to test the atomic weight may be gauged from the fact that only a few centigrams of fairly pure radium, or a few decigrams of less concentrated material, are obtained from the treatment of about 2 tons of the mineral from which it is derived.
Runge and Precht[25] have examined the spectrum of radium in a magnetic field, and have shown the existence of series analogous to those observed for calcium, barium, and strontium. These series 18are connected with the atomic weights of the elements in question, and Runge and Precht have calculated by these means that the atomic weight of radium should be 258—a number considerably greater than the number 225 obtained by Mme Curie by means of chemical analysis. Marshall Watts[26], on the other hand, using another relation between the lines of the spectrum, deduced the value obtained by Mme Curie. Runge[27] has criticised the method of deduction employed by Marshall Watts on the ground that the lines used for comparison in the different spectra were not homologous. Considering that the number found by Mme Curie agrees with that required by the periodic system, it is advisable in the present state of our knowledge to accept the experimental number rather than the one deduced by Runge and Precht from spectroscopic evidence.
There is no doubt that radium is a new element possessing remarkable physical properties. The detection and separation of this substance, existing in such minute proportions in pitchblende, has been due entirely to the characteristic property we are considering, and is the first notable triumph of the study of radio-activity. As we shall see later, the property of radio-activity can be used, not only as a means of chemical research, but also as an extraordinarily delicate method of detecting chemical changes of a very special kind.
15. Radiations from radium. On account of its enormous activity, the radiations from radium are very intense: a screen of zinc sulphide, brought near a few centigrams of radium bromide, is lighted up quite brightly in a dark room, while brilliant fluorescence is produced on a screen of platino-barium cyanide. An electroscope brought near the radium salt is discharged almost instantly, while a photographic plate is immediately affected. At a distance of one metre, a day’s exposure to the radium rays would produce a strong impression. The radiations from radium are analogous to those of uranium, and consist of three types of rays: easily absorbed, penetrating, and very penetrating. Radium also gives rise to an emanation similar to that of thorium, 19but with a very much slower rate of decay. The radium emanation retains its activity for several weeks, while that of thorium lasts only a few minutes. The emanation obtained from a few centigrams of radium illuminates a screen of zinc sulphide with great brilliancy. The very penetrating rays of radium are able to light up an X ray screen in a dark room, after passage through several centimetres of lead and several inches of iron.
As in the case of uranium or thorium, the photographic action is mainly due to the penetrating or cathodic rays. The radiographs obtained with radium are very similar to those obtained with X rays, but lack the sharpness and detail of the latter. The rays are unequally absorbed by different kinds of matter, the absorption varying approximately as the density. In photographs of the hand the bones do not stand out as in X ray photographs.
Curie and Laborde have shown that the compounds of radium possess the remarkable property of always keeping their temperature several degrees above the temperature of the surrounding air. Each gram of radium radiates an amount of energy corresponding to 100 gram-calories per hour. This and other properties of radium are discussed in detail in chapters V and XII.
16. Compounds of radium. When first prepared in the solid state, all the salts of radium—the chloride, bromide, acetate, sulphate, and carbonate—are very similar in appearance to the corresponding salts of barium, but in time they gradually become coloured. In chemical properties the salts of radium are practically the same as those of barium, with the exception that the chloride and bromide of radium are less soluble in water than the corresponding salts of barium. All the salts of radium are naturally phosphorescent. The phosphorescence of impure radium preparations is in some cases very marked.
All the radium salts possess the property of causing rapid colorations of the glass vessel which contains them. For feebly active material the colour is usually violet, for more active material a yellowish-brown, and finally black.
17. Actinium. The discovery of radium in pitchblende gave a great impetus to the chemical examination of uranium residues, and a systematic search early led to the detection of several 20new radio-active bodies. Although these show distinctive radio-active properties, so far none of them have been purified sufficiently to give a definite spectrum as in the case of radium. One of the most interesting and important of these substances was discovered by Debierne[28] while working up the uranium residues, obtained by M. and Mme Curie from the Austrian government, and was called by him actinium. This active substance is precipitated with the iron group, and appears to be very closely allied in chemical properties to thorium, though it is many thousand times more active. It is very difficult to separate from thorium and the rare earths. Debierne has made use of the following methods for partial separation:
(1) Precipitation in hot solutions, slightly acidulated with hydrochloric acid, by excess of hyposulphite of soda. The active matter is present almost entirely in the precipitate.
(2) Action of hydrofluoric acid upon the hydrates freshly precipitated, and held in suspension in water. The portion dissolved is only slightly active. By this method titanium may be separated.
(3) Precipitation of neutral nitrate solutions by oxygenated water. The precipitate carries down the active body.
(4) Precipitation of insoluble sulphates. If barium sulphate, for example, is precipitated in the solution containing the active body, the barium carries down the active matter. The thorium and actinium are freed from the barium by conversion of the sulphate into the chloride and precipitation by ammonia.
In this way Debierne has obtained a substance comparable in activity with radium. The separation, which is difficult and laborious, has not yet been carried far enough to bring out any new lines in the spectrum.
18. After the initial announcement of the discovery of actinium, several years elapsed before any definite results upon it were published by Debierne. In the meantime, Giesel[29] had independently obtained a radio-active substance from pitchblende which seemed similar in many respects to the actinium of Debierne. 21The active substance belongs to the group of cerium earths and is precipitated with them. By a succession of chemical operations, the active substance is separated mixed with lanthanum. While intensely active in comparison with thorium, the new active substance closely resembles it in radio-active properties, although, from the method of separation thorium cannot be present except in minute quantity. Giesel early observed that the substance gave off a radio-active emanation. On account of the intensity of the emanation it emits, he termed it the “emanating substance.” Recently this name has been changed to “emanium,” and under this title preparations of the active substance prepared by Giesel have been placed on the market.
Giesel found that the activity of this substance was permanent and seemed to increase during the six months’ interval after separation. In this respect it is similar to radium compounds, for the activity of radium, measured by the electric method, increases in the course of a month’s interval to four times its initial value at separation.
There can be no doubt that the “actinium” of Debierne and the “emanium” of Giesel contain the same radio-active constituent, for recent work[30] has shown that they exhibit identical radio-active properties. Each gives out easily absorbed and penetrating rays, and emits a characteristic emanation of which the rate of decay is the same for both substances. The rate of decay of the emanation is the simplest method of distinguishing actinium from thorium, which it resembles so closely in radio-active as well as in chemical properties. The emanation of actinium loses its radiating power far more rapidly than that of thorium, the time taken for the activity to fall to half value being in the two cases 3·7 seconds and 52 seconds respectively.
The rapid and continuous emission of this short-lived emanation is the most striking radio-active property possessed by actinium. In still air, the radio-active effects of this emanation are confined to a distance of a few centimetres from the active material, as it is only able to diffuse a short distance through the air before losing its radiating power. With very active preparations of actinium, 22the material appears to be surrounded by a luminous haze produced by the emanation. The radiations produce strong luminosity in some substances, for example, zinc sulphide, willemite and platinocyanide of barium. The luminosity is especially marked on screens of zinc sulphide. Much of this effect is due to the emanation, for, on gently blowing a current of air over the substance, the luminosity is displaced at once in the direction of the current. With a zinc sulphide screen, actinium shows the phenomena of “scintillations” to an even more marked degree than radium itself.
The preparations of emanium are in some cases luminous, and a spectroscopic examination of this light has shown a number of bright lines[31].
The distinctive character of the emanation of actinium, as well as of the other radio-active products to which it gives rise, coupled with the permanence of its activity, renders it very probable that actinium will prove to be a new radio-active element of very great activity. Although very active preparations of actinium have been obtained, it has not yet been found possible to free it from impurities. Consequently, no definite observations have been made on its chemical properties, and no new spectrum lines have been observed.
A more complete discussion of the radio-active and other properties of actinium is given in later chapters.
19. Polonium. Polonium was the first of the active substances obtained from pitchblende. It has been investigated in detail by its discoverer Mme Curie[32]. The pitchblende was dissolved in acid and sulphuretted hydrogen added. The precipitated sulphides contained an active substance, which, after separation of impurities, was found associated with bismuth. This active substance, which has been named polonium, is so closely allied in chemical properties to bismuth that it has so far been found impossible to effect a complete separation. Partial separation of polonium can be made by successive fractionations based on one of the following modes of procedure:
(1) Sublimation in a vacuum. The active sulphide is more 23volatile than that of bismuth. It is deposited as a black substance at those parts of the tube, where the temperature is between 250 and 300° C. In this way polonium of activity 700 times that of uranium was obtained.
(2) Precipitation of nitric acid solutions by water. The precipitated sub-nitrate is much more active than the part that remains in solution.
(3) Precipitation by sulphuretted hydrogen in a very acid hydrochloric acid solution. The precipitated sulphides are much more active than the salt which remains in solution.
For concentration of the active substance Mme Curie[33] has made use of method (2). The process is, however, very slow and tedious, and is made still more complicated by the tendency to form precipitates insoluble either in strong or weak acids. After a large number of fractionations, a small quantity of matter was obtained, enormously active compared with uranium. On examination of the substance spectroscopically, only the bismuth lines were observed. A spectroscopic examination of the active bismuth by Demarçay and by Runge and Exner has led to the discovery of no new lines. On the other hand Sir William Crookes[34] states that he found one new line in the ultra-violet, while Berndt[35], working with polonium of activity 300, observed a large number of new lines in the ultra-violet. These results await further confirmation.
The polonium prepared by Mme Curie differs from the other radio-active bodies in several particulars. In the first place the radiations include only very easily absorbable rays. The two penetrating types of radiation given out by uranium, thorium, and radium are absent. In the second place the activity does not remain constant, but diminishes continuously with the time. Mme Curie states that different preparations of polonium had somewhat different rates of decay. In some cases, the activity fell to half value in about six months, and in others, about half value in eleven months.
20. The gradual diminution of the activity of polonium with time seemed at first sight to differentiate it from such substances 24as uranium and radium, the activity of which appeared fairly permanent. This difference in behaviour is, however, one of degree rather than of kind. We shall show later that there is present in pitchblende a number of radio-active substances, the activity of which is not permanent. The time taken for these bodies to lose half of their activity varies in different cases from a few seconds to several hundreds of years. In fact, this gradual loss of activity is an essential feature of our theory of regarding the phenomena of radio-activity. No radio-active substance, left to itself, can continue to radiate indefinitely; it must ultimately lose its activity. In the case of bodies like uranium and radium, the loss of activity is so slow that no sensible alteration has been observed over a period of several years, but it can be deduced theoretically that the activity of radium will eventually decrease to half value in a period of about 1000 years, while in the case of a feebly radio-active substance like uranium, more than a 100 million years must elapse before the diminution of the activity becomes appreciable.
It may be of interest here to consider briefly the suggestions advanced at various times to account for the temporary character of the activity of polonium. Its association with bismuth led to the view that polonium was not a new active substance, but merely radio-active bismuth, that is, bismuth which in some way had been made active by admixture with radio-active bodies. It was known that a body placed in the vicinity of thorium or radium became temporarily active. The same action was supposed to take place when inactive matter was in solution with active matter. The non-active matter was supposed to acquire activity by “induction,” as it was called, in consequence of its intimate contact with the active material.
There is no proof, however, that such is the case. The evidence points rather to the conclusion that the activity is due, not to an alteration of the inactive body itself, but to an admixture with it of a very small quantity of intensely active matter. This active matter is present in pitchblende and is separated with the bismuth but differs from it in chemical properties.
The subject cannot be considered with advantage at this stage, but will be discussed later in detail in chapter XI. It will 25there be shown that polonium, that is, the radio-active constituent mixed with the bismuth, is a distinct chemical substance, which is allied in chemical properties to bismuth, but possesses some distinct analytical properties which allow of a partial separation from it.
The polonium, if obtained in a pure state, should initially be several hundred times as active as pure radium. This activity, however, is not permanent; it decays with the time, falling to half value in about six months.
The absence of any new lines in the spectrum of radio-active bismuth is to be expected, for, even in the most active bismuth prepared, the active matter exists in a very small proportion.
21. The discussion of the nature of polonium was renewed by the discovery of Marckwald[36] that a substance similar to polonium can be separated from pitchblende; the activity of this substance, he stated, did not decay appreciably with the time. The method of separation from the bismuth chloride solution, obtained from uranium residues, was very simple. A rod of bismuth or antimony, dipped in the active solution, rapidly became coated with a black deposit which was intensely active. This process was continued until the whole of the activity was removed from the solution. The active deposit gave out only easily absorbed rays, and in that respect resembled the polonium of Mme Curie.
The active substance was found to consist mainly of tellurium, and for this reason Marckwald gave it the name of radio-tellurium. In later work, however, Marckwald[37] has shown that the active constituent has no connection with tellurium, but can always be separated completely from it by a simple chemical process.
In order to obtain a large amount of the active substance, 2000 kilos. of pitchblende were worked up. This yielded 6 kilos. of bismuth oxychloride, and from this was separated 1·5 grams of radio-tellurium. The tellurium present was precipitated from a hydrochloric acid solution by hydrazine hydrochloride. The precipitated tellurium still showed some activity, but this was removed by repeating the process. The active matter then 26remained in the filtrate, and, after evaporation, the addition of a few drops of stannous chloride caused a small quantity of a dark precipitate which was intensely active. This was collected on a filter and weighed only 4 milligrams.
When plates of copper, tin or bismuth were dipped into an hydrochloric acid solution of this active substance, the plates were found to be covered with a very finely divided deposit. These plates were intensely active, and produced marked photographic and phosphorescent action. As an illustration of the enormous activity of this deposit, Marckwald stated that a precipitate of ¹⁄₁₀₀ milligram on a copper plate, 4 square centimetres in area, illuminated a zinc sulphide screen so brightly that it could be seen by an audience of several hundred people.
The active substance of Marckwald is very closely allied in chemical and radio-active properties to the polonium of Mme Curie. Both active substances are separated with bismuth and both give out only easily absorbed rays. The penetrating rays, such as are given out by uranium, radium or thorium, are completely absent.
There has been a considerable amount of discussion as to whether the active substance obtained by Marckwald is identical with that present in the polonium of Mme Curie. Marckwald stated that his active substance did not sensibly diminish in activity in the course of six months, but it is doubtful whether the method of measurement used was sufficiently precise.
The writer has found that radio-tellurium of moderate activity, prepared after Marckwald’s method and sold by Dr Sthamer of Hamburg, undoubtedly loses its activity with time. The radio-tellurium is obtained in the form of a thin radio-active deposit on a polished bismuth rod or plate. A bismuth rod was found to have lost half its activity in about 150 days, and a similar result has been recorded by other observers.
The two substances are thus similar in both radio-active and chemical properties, and there can be no reasonable doubt that the active constituent present in each case is the same. The evidence is discussed in detail in chapter XI and it will there be shown that the active substance present in the radio-tellurium of Marckwald is a slow transformation product of radium.
27 22. Radio-active lead. Several observers early noticed that the lead separated from pitchblende showed strong radio-active properties, but considerable difference of opinion was expressed in regard to the permanence of its activity. Elster and Geitel[38] found that lead sulphate obtained from pitchblende was very active, but they considered that the activity was probably due to an admixture of radium or polonium with the lead, and, by suitable chemical treatment, the lead sulphate was obtained in an inactive state. Giesel[39] also separated some radio-active lead but found that its activity diminished with the time. On the other hand, Hofmann and Strauss[40] obtained lead from pitchblende whose activity seemed fairly permanent. They state that the radio-active lead resembled ordinary lead in most of its reactions, but showed differences in the behaviour of the sulphide and sulphate. The sulphate was found to be strongly phosphorescent. These results of Hofmann and Strauss were subjected at the time of their publication to considerable criticism, and there is no doubt that the lead itself is not radio-active but contains a small quantity of radio-active matter which is separated with it. In later work[41], it has been shown that radio-lead contains several radio-active constituents which can be removed temporarily from it by suitable chemical methods.
There can be no doubt that the lead separated from pitchblende by certain methods does show considerable activity and that this activity is fairly permanent. The radio-active changes occurring in radio-lead are complicated and cannot be discussed with advantage at this stage, but will be considered in detail in chapter XI. It will there be shown that the primary constituent present in lead is a slow transformation product of radium. This substance then slowly changes into the active constituent present in polonium, which gives out only easily absorbed rays.
This polonium can be separated temporarily from the lead by suitable chemical methods, but the radio-lead still continues to produce polonium, so that a fresh supply may be obtained 28from it, provided an interval of several months is allowed to elapse.
It will be calculated later that in all probability the radio-lead would lose half of its activity in an interval of 40 years.
The constituent present in radio-lead has not yet been separated, but it will be shown that, in the pure state, it should have an activity considerably greater than that of radium itself. Sufficient attention has not yet been paid to this substance, for, separated in a pure state, it should be as useful scientifically as radium. In addition, since it is the parent of polonium, it should be possible to obtain from it at any time a supply of very active polonium, in the same way that a supply of the radium emanation can be obtained at intervals from radium.
Hofmann and Strauss have observed a peculiar action of the cathode rays on the active lead sulphate separated by them. They state that the activity diminishes with time, but is recovered by exposure of the lead for a short time to the action of cathode rays. No such action is shown by the active lead sulphide. This effect is due most probably to the action of the cathode rays in causing a strong phosphorescence of the lead sulphate and has nothing to do with the radio-activity proper of the substance.
23. Is thorium a radio-active element? The similarity of the chemical properties of actinium and thorium has led to the suggestion at different times that the activity of thorium is not due to thorium itself, but to the presence of a slight trace of actinium. In view of the difference in the rate of decay of the emanations of thorium and actinium, this position is not tenable. If the activity of thorium were due to actinium, the two emanations, as well as the other products obtained from these substances, should have identical rates of decay. Since there is not the slightest evidence that the rate of decay of activity of the various products can be altered by chemical or physical agencies, we may conclude with confidence that whatever radio-active substance is responsible for the activity of thorium, it certainly is not actinium. This difference in the rate of decay of the active products is of far more weight in deciding the question whether two bodies contain the same radio-active constituent than differences in chemical 29behaviour, for it is quite probable that the active material in each case may exist only in minute quantity in the matter under examination, and, under such conditions, a direct chemical examination in the first place is of little value.
Recent work of Hofmann and Zerban and of Baskerville, however, certainly tends to show that the element thorium is itself non-radio-active, and that the radio-activity observed in ordinary thorium compounds is due to the admixture with it of an unknown radio-active element. Hofmann and Zerban[42] made a systematic examination of the radio-activity of thorium obtained from different mineral sources. They found generally that thorium, obtained from minerals containing a large percentage of uranium, were more active than those obtained from minerals nearly free from uranium. This indicates that the radio-activity observed in thorium may possibly be due to a transformation product of uranium which is closely allied chemically to thorium and is always separated with it. A small quantity of thorium obtained from the mineral gadolinite was found by Hofmann to be almost inactive, whether tested by the electric or by the photographic method. Later Baskerville and Zerban[43] found that thorium obtained from a Brazilian mineral was practically devoid of activity.
In this connection the recent work of Baskerville on the complexity of ordinary thorium is of interest. By special chemical methods, he succeeded in separating two new and distinct substances from thorium, which he has named carolinium and berzelium. Both of these substances are strongly radio-active, and it thus seems probable that the active constituent observed in ordinary thorium may be due to one of these elements.
If, as we have suggested, thorium itself is not active, it is certainly a matter of surprise that ordinary commercial thorium and the purest chemical preparations show about the same activity. Such a result indicates that the methods of purification have not removed any of the radio-active constituent originally present.
Whatever the radio-active constituent in thorium may ultimately prove to be, it is undoubtedly not radium nor actinium nor any of the known radio-active substances.
30In later chapters, the radio-activity of thorium will, for simplicity, be discussed on the assumption that thorium is itself a radio-active element. The analysis of the changes which occur will thus not refer to thorium itself but to the primary radio-active substance usually found associated with it. The conclusions to be drawn from an examination of the radio-active processes are for the most part independent of whether thorium is itself radio-active or whether the radio-activity is due to an unknown element. If thorium is not radio-active itself, it is not possible to draw any conclusions upon the question of the duration of the primary radio-activity associated with it. Such a deduction cannot be made until the quantity of the radio-active element present in thorium has been definitely determined.
24. If elements heavier than uranium exist, it is probable that they will be radio-active. The extreme delicacy of radio-activity as a means of chemical analysis would enable such elements to be recognized even if present in infinitesimal quantities. It is probable that considerably more than the three or four radio-elements at present recognized exist in minute quantity, and that the number at present known will be augmented in the future. In the first stage of the search, a purely chemical examination is of little value, for it is not probable that the new element should exist in sufficient quantity to be detected by chemical or spectroscopic analysis. The main criteria of importance are the existence or absence of distinctive radiations or emanations, and the permanence of the radio-activity. The discovery of a radio-active emanation with a rate of decay different from those already known would afford strong evidence that a new radio-active body was present. The presence of either thorium or radium in matter can very readily be detected by observing the rate of decay of the emanations given out by them. When once the existence of a new radio-element has been inferred by an examination of its radio-active properties, chemical methods of separation can be devised, the radiating or emanating property being used as a guide in qualitative and quantitative analysis.
25. Ionization of gases by radiation. The most important property possessed by the radiations from radio-active bodies is their power of discharging bodies whether positively or negatively electrified. As this property has been made the basis of a method for an accurate quantitative analysis and comparison of the radiations, the variation of the rate of discharge under different conditions and the processes underlying it will be considered in some detail.
In order to explain the similar discharging power of Röntgen rays, the theory[44] has been put forward that the rays produce positively and negatively charged carriers throughout the volume of the gas surrounding the charged body, and that the rate of production is proportional to the intensity of the radiation. These carriers, or ions[45] as they have been termed, move with a uniform velocity through the gas under a constant electric field, and their velocity varies directly as the strength of the field.
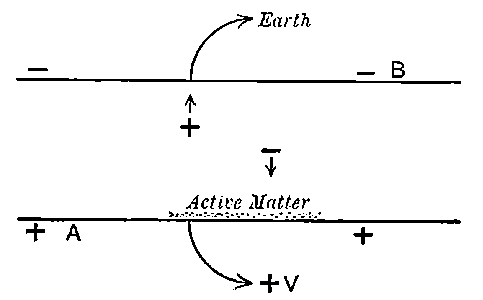
Fig. 1.
Suppose we have a gas between two metal plates A and B (Fig. 1) exposed to the radiation, and that the plates are kept at a constant difference of potential. A definite number of ions will be produced per second by the radiation, and the number 32produced will depend in general upon the nature and pressure of the gas. In the electric field the positive ions travel towards the negative plate, and the negative ions towards the positive, and consequently a current will pass through the gas. Some of the ions will also recombine, the rate of recombination being proportional to the square of the number present. For a given intensity of radiation, the current passing through the gas will increase at first with the potential difference between the plates, but it will reach a limit when all the ions are removed by the electric field before any recombination occurs.
This theory accounts also for all the characteristic properties of gases made conducting by the rays from active substances, though there are certain differences observed between the conductivity phenomena produced by active substances and by X rays. These differences are for the most part the result of unequal absorption of the two types of rays. Unlike Röntgen rays, a large proportion of the radiation from active bodies consists of rays which are absorbed in their passage through a few centimetres of air. The ionization of the gas is thus not uniform, but falls off rapidly with increase of distance from the active substance.
26. Variation of the current with voltage. Suppose that a layer of radio-active matter is spread uniformly on the lower of two horizontal plates A and B (Fig. 1). The lower plate A is connected with one pole of a battery of cells the other pole of which is connected with earth. The plate B is connected with one pair of quadrants of an electrometer, the other pair being connected with earth.
The current[46] between the plates, determined by the rate of movement of the electrometer needle, is observed at first to increase rapidly with the voltage, then more slowly, finally reaching a value which increases very slightly with a large increase in the voltage. This, as we have indicated, is simply explained on the ionization theory.
The radiation produces ions at a constant rate, and, before the electric field is applied, the number per unit volume increases 33until the rate of production of fresh ions is exactly balanced by the recombination of the ions already produced. On application of a small electric field, the positive ions travel to the negative electrode and the negative to the positive.
Since the velocity of the ions between the plates is directly proportional to the strength of the electric field, in a weak field the ions take so long to travel between the electrodes that most of them recombine on the way.
The current observed is consequently small. With increase of the voltage there is an increase of speed of the ions and a smaller number recombine. The current consequently increases, and will reach a maximum value when the electric field is sufficiently strong to remove all the ions before appreciable recombination has occurred. The value of the current will then remain constant even though the voltage is largely increased.
This maximum current will be called the “saturation” current, and the value of the potential difference required to give this maximum current, the “saturation P.D.”[47]
The general shape of the current-voltage curve is shown in Fig. 2, where the ordinates represent current and the abscissae volts.
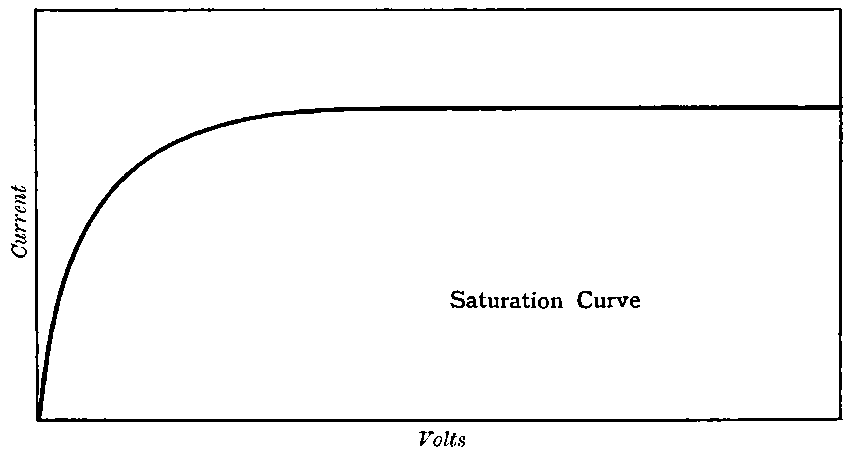
Fig. 2.
34Although the variation of the current with voltage depends only on the velocity of the ions and their rate of recombination, the full mathematical analysis is intricate, and the equations, expressing the relation between current and voltage, are only integrable for the case of uniform ionization. The question is complicated by the inequality in the velocity of the ions and by the disturbance of the potential gradient between the plates by the movement of the ions. J. J. Thomson[48] has worked out the case for uniform production of ions between two parallel plates, and has found that the relation between the current i and the potential difference V applied is expressed by
where A and B are constants for a definite intensity of radiation and a definite distance between the plates.
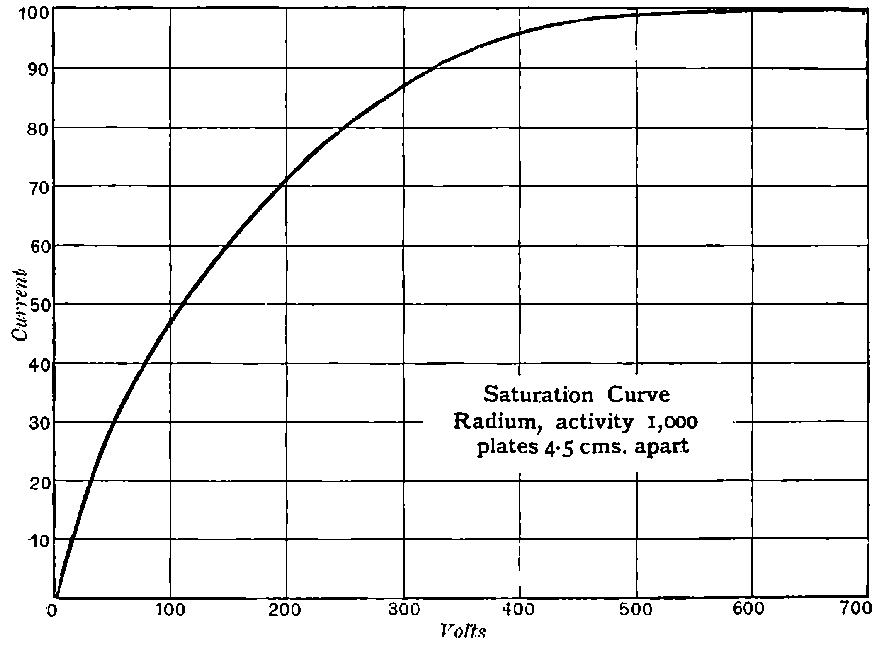
Fig. 3.
In certain cases of unsymmetrical ionization, which arise in the study of the radiations from active bodies, the relation between current and voltage is very different from that expressed by 35the above equation. Some of these cases will be considered in section 47.
27. The general shape of the current-voltage curves for gases exposed to the radiations from active bodies is shown in Fig. 3.
This curve was obtained for ·45 grams of impure radium chloride, of activity 1000 times that of uranium, spread over an area of 33 sq. cms. on the lower of two large parallel plates, 4·5 cms. apart. The maximum value of the current observed, which is taken as 100, was 1·2 × 10-8 amperes, the current for low voltages was nearly proportional to the voltage, and about 600 volts between the plates was required to ensure approximate saturation.
In dealing with slightly active bodies like uranium or thorium, approximate saturation is obtained for much lower voltages. Tables I. and II. show the results for the current between two parallel plates distant 0·5 cms. and 2·5 cms. apart respectively, when one plate was covered with a thin uniform layer of uranium oxide.
| Volts | Current |
|---|---|
| ·125 | 18 |
| ·25 | 36 |
| ·5 | 55 |
| 1 | 67 |
| 2 | 72 |
| 4 | 79 |
| 8 | 85 |
| 16 | 88 |
| 100 | 94 |
| 335 | 100 |
| Volts | Current |
|---|---|
| ·5 | 7·3 |
| 1 | 14 |
| 2 | 27 |
| 4 | 47 |
| 8 | 64 |
| 16 | 73 |
| 37·5 | 81 |
| 112 | 90 |
| 375 | 97 |
| 800 | 100 |
The results are shown graphically in Fig. 4.
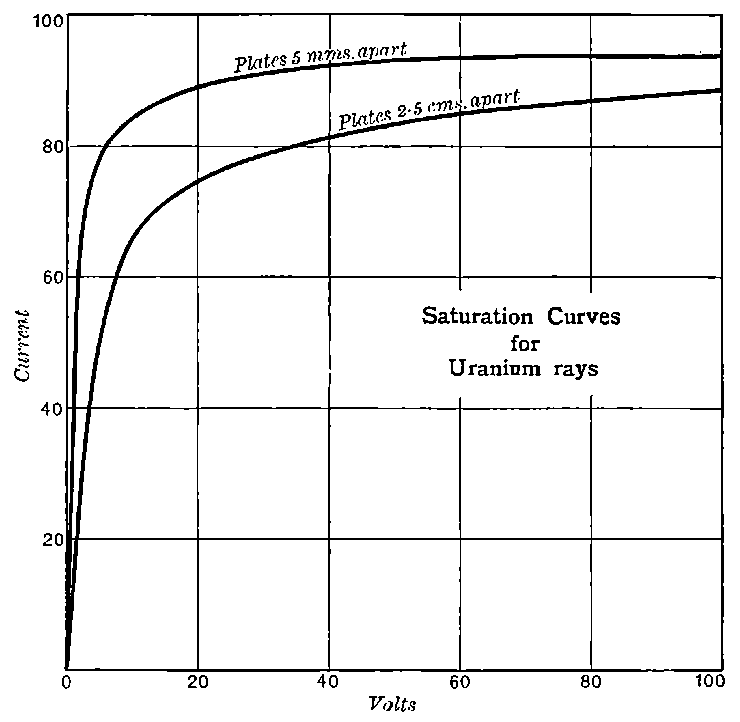
Fig. 4.
From the above tables it is seen that the current at first increases nearly in proportion to the voltage. There is no evidence of complete saturation, although the current increases very slowly for large increases of voltage. For example, in Table I. a change of voltage from ·125 to ·25 volts increases the current from 18 to 36% of the maximum, while a change of voltage from 100 to 335 volts increases the current only 6%. The variation of the current per volt (assumed uniform between the range of voltages considered) is thus about 5000 times greater for the former change.
36Taking into consideration the early part of the curves, the current does not reach a practical maximum as soon as would be expected on the simple ionization theory. It seems probable that the slow increase with the large voltages is due either to an action of the electric field on the rate of production of ions, or to the difficulty of removing the ions produced near the surface of the uranium before recombination. It is possible that the presence of a strong electric field may assist in the separation of ions which otherwise would not initially escape from the sphere of one another’s attraction. From the data obtained by Townsend for the conditions of production of fresh ions at low pressures by the movement of ions through the gas, it seems that the increase of current cannot be ascribed to an action of the moving ions in the further ionization of the gas.
28. The equation expressing the relation between the current and the voltage is very complicated even in the case of a uniform rate of production of ions between the plates. An approximate 37theory, which is of utility in interpreting the experimental results, can however be simply deduced if the disturbance of the potential gradient is disregarded, and the ionization assumed uniform between the plates.
Suppose that the ions are produced at a constant rate q per cubic centimetre per second in the gas between parallel plates distant l cms. from each other. When no electric field is applied, the number N present per c.c., when there is equilibrium between the rates of production and recombination, is given by
where α is a constant.
If a small potential difference V is applied, which gives only a small fraction of the maximum current, and consequently has not much effect on the value of N, the current i per sq. cm. of the plate, is given by
where u is the sum of the velocity of the ions for unit potential gradient, and e is the charge carried by an ion.
is the velocity of the ions in the electric field of strength
The number of ions produced per second in a prism of length l and unit area of cross-section is ql. The maximum or saturation current I per sq. cm. of the plate is obtained when all of these ions are removed to the electrodes before any recombination has occurred.
Thus
and
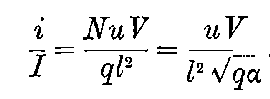
This equation expresses the fact previously noted that, for small voltages, the current i is proportional to V.
Let
then
38Now the greater the value of V required to obtain a given value of ρ (supposed small compared with unity), the greater the potential required to produce saturation.
It thus follows from the equation that:
(1) For a given intensity of radiation, the saturation P.D. increases with the distance between the plates. In the equation, for small values of ρ, V varies as l2. This is found to be the case for uniform ionization, but it only holds approximately for non-uniform ionization.
(2) For a given distance between the plates, the saturation P.D. is greater, the greater the intensity of ionization between the plates. This is found to be the case for the ionization produced by radio-active substances. With a very active substance like radium, the ionization produced is so intense that very large voltages are required to produce approximate saturation. On the other hand, only a fraction of a volt per cm. is necessary to produce saturation in a gas where the ionization is very slight, for example, in the case of the natural ionization observed in a closed vessel, where no radio-active substances are present.
For a given intensity of radiation, the saturation P.D. decreases rapidly with the lowering of the pressure of the gas. This is due to two causes operating in the same direction, viz. a decrease in the intensity of the ionization and an increase in the velocity of the ions. The ionization varies directly as the pressure, while the velocity varies inversely as the pressure. This will obviously have the effect of causing more rapid saturation, since the rate of recombination is slower and the time taken for the ions to travel between the electrodes is less.
The saturation curves observed for the gases hydrogen and carbon dioxide[49] are very similar in shape to those obtained for air. For a given intensity of radiation, saturation is more readily obtained in hydrogen than in air, since the ionization is less than in air while the velocity of the ions is greater. Carbon dioxide on the other hand requires a greater P.D. to produce saturation than does air, since the ionization is more intense and the velocity of the ions less than in air.
39 29. Townsend[50] has shown that, for low pressures, the variation of the current with the voltage is very different from that observed at atmospheric pressure. If the increase of current with the voltage is determined for gases, exposed to Röntgen rays, at a pressure of about 1 mm. of mercury, it is found that for small voltages the ordinary saturation curve is obtained; but when the voltage applied increases beyond a certain value, depending on the pressure and nature of the gas and the distance between the electrodes, the current commences to increase slowly at first but very rapidly as the voltage is raised to the sparking value. The general shape of the current curve is shown in Fig. 5.
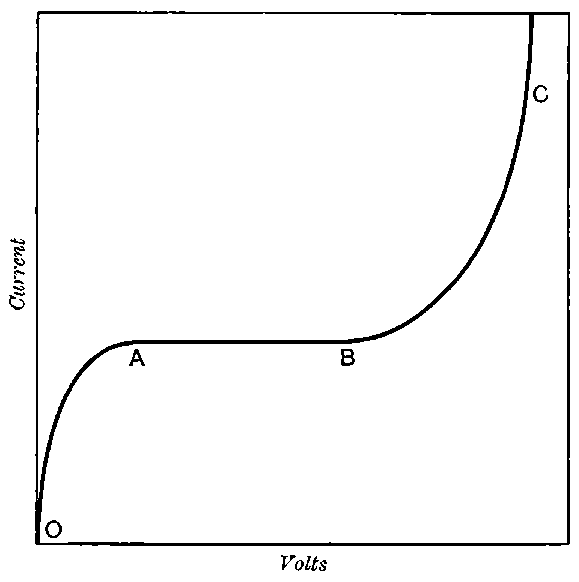
Fig. 5.
The portion OAB of the curve corresponds to the ordinary saturation curve. At the point B the current commences to increase. This increase of current has been shown to be due to the action of the negative ions at low pressures in producing fresh ions by collision with the molecules in their path. The increase of current is not observed in air at a pressure above 30 mms. until the P.D. is increased nearly to the value required to produce a spark. This production of ions by collision is considered in more detail in section 41.
40 30. Rate of recombination of the ions. A gas ionized by the radiation preserves its conducting power for some time after it is removed from the presence of the active body. A current of air blown over an active body will thus discharge an electrified body some distance away. The duration of this after conductivity can be examined very conveniently in an apparatus similar to that shown in Fig. 6.
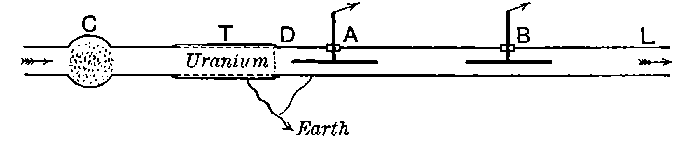
Fig. 6.
A dry current of air or any other gas is passed at a constant rate through a long metal tube TL. After passing through a quantity of cotton-wool to remove dust particles, the current of air passes over a vessel T containing a radio-active body such as uranium, which does not give off a radio-active emanation. By means of insulated electrodes A and B, charged to a suitable potential, the current between the tube and one of these electrodes can be tested at various points along the tube.
A gauze screen, placed over the cross-section of the tube at D, serves to prevent any direct action of the electric field in abstracting ions from the neighbourhood of T.
If the electric field is sufficiently strong, all the ions travel in to the electrodes at A, and no current is observed at the electrode B. If the current is observed successively at different distances along the tube, all the electrodes except the one under consideration being connected to earth, it is found that the current diminishes with the distance from the active body. If the tube is of fairly wide bore, the loss of the ions due to diffusion is small, and the decrease in conductivity of the gas is due to recombination of the ions alone.
On the ionization theory, the number dn of ions per unit volume which recombine in the time dt is proportional to the square of the number present. Thus
where α is a constant.
41Integrating this equation,
if N is the initial number of ions, and n the number after a time t.
The experimental results obtained[51] have been shown to agree very well with this equation.
In an experiment similar to that illustrated in Fig. 6, using uranium oxide as a source of ionization, it was found that half the number of ions present in the gas recombined in 2·4 seconds, and that at the end of 8 seconds one-fourth of the ions were still uncombined.
Since the rate of recombination is proportional to the square of the number present, the time taken for half of the ions present in the gas to recombine decreases very rapidly with the intensity of the ionization. If radium is used, the ionization is so intense that the rate of recombination is extremely rapid. It is on account of this rapidity of recombination that large voltages are necessary to produce saturation in the gases exposed to very active preparations of radium.
The value of α, which may be termed the coefficient of recombination, has been determined in absolute measure by Townsend[52], McClung[53] and Langevin[54] by different experimental methods but with very concordant results. Suppose, for example, with the apparatus of Fig. 6, the time T, taken for half the ions to recombine after passing by the electrode A, has been determined experimentally. Then
where N is the number of ions per c.c. present at A. If the saturation current i is determined at the electrode A, i = NVe, where e is the charge on an ion and V is the volume of uniformly ionized gas carried by the electrode A per second. Then
The following table shows the value of α obtained for different gases.
| Gas | Townsend | McClung | Langevin |
|---|---|---|---|
| Air | 3420 × e | 3384 × e | 3200 × e |
| Carbon Dioxide | 3500 × e | 3492 × e | 3400 × e |
| Hydrogen | 3020 × e |
The latest determination of the value of e (see section 36) is 3·4 × 10-10 E.S. units; thus α = 1·1 × 10-6.
Using this value, it can readily be shown from the equation of recombination that, if 106 ions are present per c.c., half of them recombine in about 0·9 sec. and 99% in 90 secs.
McClung (loc. cit.) showed that the value of α was approximately independent of the pressure between ·125 and three atmospheres. In later observations, Langevin has found that the value of α decreases rapidly when the pressure is lowered below the limits used by McClung.
31. In experiments on recombination it is essential that the gas should be free from dust or other suspended particles. In dusty air, the rate of recombination is much more rapid than in dust-free air, as the ions diffuse rapidly to the comparatively large dust particles distributed throughout the gas. The effect of the suspension of small particles in a conducting gas is very well illustrated by an experiment of Owens[55]. If tobacco smoke is blown between two parallel plates as in Fig. 1, the current at once diminishes to a small fraction of its former value, although a P.D. is applied sufficient to produce saturation under ordinary conditions. A much larger voltage is then necessary to produce saturation. If the smoke particles are removed by a stream of air, the current returns at once to its original value.
32. Mobility of the ions. Determinations of the mobility of the ions, i.e. the velocity of the ions under a potential gradient of 1 volt per cm., have been made by Rutherford[56], Zeleny[57], and Langevin[58] for gases exposed to Röntgen rays. Although widely different methods have been employed, the results have been very concordant, and fully support the view that the ions move with a 43velocity proportional to the strength of the field. On the application of an electric field, the ions almost instantly attain the velocity corresponding to the field and then move with a uniform speed.
Zeleny[59] first drew attention to the fact that the positive and negative ions had different velocities. The velocity of the negative ion is always greater than that of the positive, and varies with the amount of water vapour present in the gas.
The results, previously discussed, of the variation of the current with voltage and of the rate of recombination of the ions do not of themselves imply that the ions produced in gases by the radiations from active bodies are of the same size as those produced by Röntgen rays under similar conditions. They merely show that the conductivity under various conditions can be satisfactorily explained by the view that charged ions are produced throughout the volume of the gas. The same general relations would be observed if the ions differed considerably in size and velocity from those produced by Röntgen rays. The most satisfactory method of determining whether the ions are identical in the two cases is to determine the velocity of the ions under similar conditions.
In order to compare the velocity of the ions[60], the writer has used an apparatus similar to that shown in Fig. 6 on p. 40.
The ions were carried with a rapid constant stream of air past the charged electrode A, and the conductivity of the gas tested immediately afterwards at an electrode B, which was placed close to A. The insulated electrodes A and B were fixed centrally in the metal tube L, which was connected with earth.
For convenience of calculation, it is assumed that the electric field between the cylinders is the same as if the cylinders were infinitely long.
Let a and b be the radii of the electrode A, and of the tube L respectively, and let V = potential of A.
The electromotive intensity X (without regard to sign) at a distance r from the centre of the tube is given by
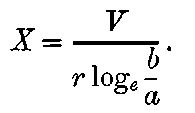
44Let u1 and u2 be the velocities of the positive and negative ions for a potential gradient of 1 volt per cm. If the velocity is proportional to the electric force at any point, the distance dr traversed by the negative ion in the time dt is given by
dr = Xu2 dt,
or
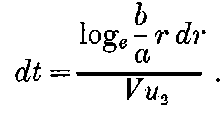
Let r2 be the greatest distance measured from the axis of the tube from which the negative ion can just reach the electrode A in the time t taken for the air to pass along the electrode.
Then
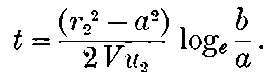
If ρ2 be the ratio of the number of the negative ions that reach the electrode A to the total number passing by, then
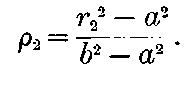
Therefore
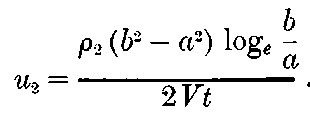
Equation 1.
Similarly the ratio ρ1 of the number of positive ions that give up their charge to the external cylinder to the total number of positive ions is given by
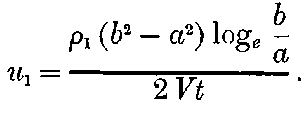
In the above equations it is assumed that the current of air is uniform over the cross-section of the tube, and that the ions are uniformly distributed over the cross-section; also, that the movement of the ions does not appreciably disturb the electric field. Since the value of t can be calculated from the velocity of the current of air and the length of the electrode, the values of the velocities of the ions under unit potential gradient can at once be determined.
The equation (1) shows that ρ2 is proportional to V,—i.e. that 45the rate of discharge of the electrode A varies directly as the potential of A, provided that the value of V is not large enough to remove all the ions from the gas as it passes by the electrode. This was found experimentally to be the case.
In the comparison of the velocities, the potential V was adjusted to such a value that ρ2 was about one half, when uranium oxide was placed in the tube at L. The active substance was then removed, and an aluminium cylinder substituted for the brass tube. X rays were allowed to fall on the centre of this aluminium cylinder, and the strength of the rays adjusted to give about the same conductivity to the gas as the uranium had done. Under these conditions the value of ρ2 was found to be the same as for the first experiment.
This experiment shows conclusively that the ions produced by Röntgen rays and by uranium move with the same velocity and are probably identical in all respects. The method described above is not very suitable for an accurate determination of the velocities, but gave values for the positive ions of about 1·4 cms. per second per volt per centimetre, and slightly greater values for the negative ions.
33. The most accurate determinations of the mobility of the ions produced by Röntgen rays have been made by Zeleny[61] and Langevin[62]. Zeleny used a method similar in principle to that explained above. His results are shown in the following table, where K1 is the mobility of the positive ion and K2 that of the negative ion.
| Gas | K1 | K2 | K2/K1 | Temperature |
|---|---|---|---|---|
| Air, dry | 1·36 | 1·87 | 1·375 | 13°·5 C. |
| „ moist | 1·37 | 1·51 | 1·10 | 14° |
| Oxygen, dry | 1·36 | 1·80 | 1·32 | 17° |
| „ moist | 1·29 | 1·52 | 1·18 | 16° |
| Carbon dioxide, dry | 0·76 | 0·81 | 1·07 | 17°·5 |
| „ „ moist | 0·81 | 0·75 | 0·915 | 17° |
| Hydrogen, dry | 6·70 | 7·95 | 1·15 | 20° |
| „ moist | 5·30 | 5·60 | 1·05 | 20° |
46Langevin determined the velocity of the ions by a direct method in which the time taken for the ion to travel over a known distance was observed.
The following table shows the comparative values obtained for air and carbon dioxide.
| Air K1 | Air K2 | Air K2/K1 | CO2 K1 | CO2 K2 | CO2 K2/K1 | |
|---|---|---|---|---|---|---|
| Direct method (Langevin) | 1·40 | 1·70 | 1·22 | 0·86 | 0·90 | 1·05 |
| Current of gas (Zeleny) | 1·36 | 1·87 | 1·375 | 0·76 | 0·81 | 1·07 |
These results show that for all gases except CO2, there is a marked increase in the velocity of the negative ion with the dryness of the gas, and that, even in moist gases, the velocity of the negative ions is always greater than that of the positive ions. The velocity of the positive ion is not much affected by the presence of moisture in the gas.
The velocity of the ions varies inversely as the pressure of the gas. This has been shown by Rutherford[63] for the negative ions produced by ultra-violet light falling on a negatively charged surface, and later by Langevin[64] for both the positive and negative ions produced by Röntgen rays. Langevin has shown that the velocity of the positive ion increases more slowly with the diminution of pressure than that of the negative ion. It appears as if the negative ion, especially at pressures of about 10 mm. of mercury, begins to diminish in size.
34. Condensation experiments. Some experiments will now be described which have verified in a direct way the theory that the conductivity produced in gases by the various types of radiation is due to the production of charged ions throughout the volume of the gas. Under certain conditions, the ions form nuclei for the condensation of water, and this property allows us to show the presence of the individual ions in the gas, and also to count the number present.
It has long been known that, if air saturated with water-vapour be suddenly expanded, a cloud of small globules of water is formed. These drops are formed round the dust particles present in the gas, 47which act as nuclei for the condensation of water around them. The experiments of R. von Helmholtz and Richarz[65] had shown that chemical reactions, for example the combustion of flames, taking place in the neighbourhood, affected the condensation of a steam-jet. Lenard showed that a similar action was produced when ultra-violet light fell on a negatively charged zinc surface placed near the steam-jet. These results suggested that the presence of electric charges in the gas facilitated condensation.
A very complete study of the conditions of condensation of water on nuclei has been made by C. T. R. Wilson[66]. An apparatus was constructed which allowed a very sudden expansion of the air over a wide range of pressure. The amount of condensation was observed in a small glass vessel. A beam of light was passed into the apparatus which allowed the drops formed to be readily observed by the eye.
Preliminary small expansions caused a condensation of the water round the dust nuclei present in the air. These dust nuclei were removed by allowing the drops to settle. After a number of successive small expansions, the air was completely freed from dust, so that no condensation was produced.
Let v1 = initial volume of the gas in the vessel, v2 = volume after expansion.
If v2/v1 < 1·25 no condensation is produced in dust-free air. If however v2/v1 > 1·25 and < 1·38, a few drops appear. This number is roughly constant until v2/v1 = 1·38, when the number suddenly increases and a very dense cloud of fine drops is produced.
If the radiation from an X ray tube or a radio-active substance is now passed into the condensation vessel, a new series of phenomena is observed. As before, if v2/v1 < 1·25 no drops are formed, but if v2/v1 = 1·25 there is a sudden production of a cloud. The water drops of which this cloud is formed are finer and more numerous the 48greater the intensity of the rays. The point at which condensation begins is very marked, and a slight variation of the amount of expansion causes either a dense cloud or no cloud at all.
It now remains to be shown that the formation of a cloud by the action of the rays is due to the productions of ions in the gas. If the expansion vessel is provided with two parallel plates between which an electric field can be applied, it is seen that the number of drops, formed by the expansion with the rays acting, decreases with increase of the electric field. The stronger the field the smaller the number of drops formed. This result is to be expected if the ions are the centres of condensation; for in a strong electric field the ions are carried at once to the electrodes, and thus disappear from the gas. If no electric field is acting, a cloud can be produced some time after the rays have been cut off; but if a strong electric field is applied, under the same conditions, no cloud is formed. This is in agreement with experiments showing the time required for the ions to disappear by recombination. In addition it can be shown that each one of the fine drops carries an electric charge and can be made to move in a strong uniform electric field.
The small number of drops produced without the action of the rays when v2/v1 > 1·25 is due to a very slight natural ionization of the gas. That this ionization exists has been clearly shown by electrical methods (section 284).
The evidence is thus complete that the ions themselves serve as centres for the condensation of water around them. These experiments show conclusively that the passage of electricity through a gas is due to the presence of charged ions distributed throughout the volume of the gas, and verify in a remarkable way the hypothesis of the discontinuous structure of the electric charges carried by matter.
This property of the ions of acting as nuclei of condensation gives a very delicate method of detecting the presence of ions in the gas. If only an ion or two is present per c.c., their presence after expansion is at once observed by the drops formed. In this way the ionization due to a small quantity of uranium held a yard away from the condensation vessel is at once made manifest.
4935. Difference between the positive and negative ions. In the course of experiments to determine the charge carried by an ion, J. J. Thomson[67] observed that the cloud formed under the influence of X rays increased in density when the expansion was about 1·31, and suggested in explanation that the positive and negative ions had different condensation points.
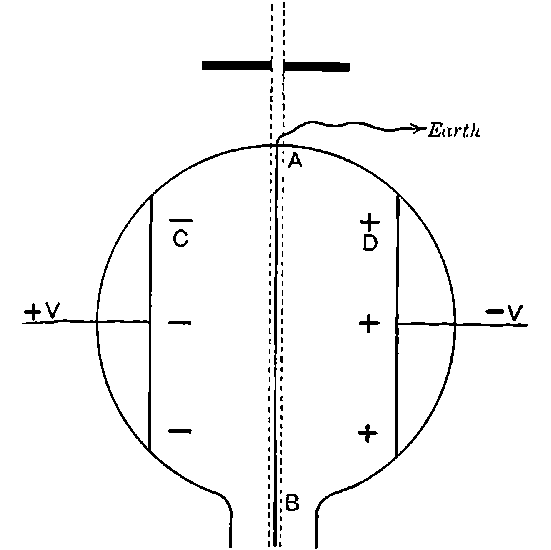
Fig. 7.
This difference in behaviour of the positive and negative ions was investigated in detail by C. T. R. Wilson[68] in the following way. X rays were made to pass in a narrow beam on either side of a plate AB (Fig. 7) dividing the condensation vessel into two equal parts. The opposite poles of a battery of cells were connected with two parallel plates C and D, placed symmetrically with regard to A. The middle point of the battery and the plate A were connected with earth. If the plate C is positively charged, the ions in the space CA at a short distance from A are all negative in sign. Those to the right are all positive. It was found that condensation occurred only for the negative ions in AC when v2/v1 = 1·25 but did not occur in AD for the positive ions until v2/v1 = 1·31.
50Thus the negative acts more readily than the positive ion as a centre of condensation. The greater effect of the negative ion in causing condensation has been suggested as an explanation of the positive charge always observed in the upper atmosphere. The negative ions under certain conditions become centres for the formation of small drops of water and are removed to the earth by the action of gravity, while the positive ions remain suspended.
With the apparatus described above, it has been shown that the positive and negative ions are equal in number. If the expansion is large enough to ensure condensation on both ions, the drops formed on the right and left of the vessel in Fig. 7 are equal in number and fall at the same rate, i.e. are equal in size.
Since the ions are produced in equal numbers from a gas electrically neutral, this experiment shows that the charges on positive and negative ions are equal in value but opposite in sign.
36. Charge carried by an ion. For a known sudden expansion of a gas saturated with water vapour, the amount of water precipitated on the ions can be calculated readily. The size of the drops can be determined by observing the rate at which the cloud settles under the action of gravity. From Stokes’ equation, the terminal velocity u of a small sphere of radius r and density d falling through a gas of which the coefficient of viscosity is μ is given by
where g is the acceleration due to gravity. The radius of the drop and consequently the weight of water in each drop can thus be determined. Since the total weight of water precipitated is known, the number of drops present is obtained at once.
This method has been used by J. J. Thomson[69] to determine the charge carried by an ion. If the expansion exceeds the value 1·31, both positive and negative ions become centres of condensation. From the rate of fall it can be shown that approximately the drops are all of the same size.
51The condensation vessel was similar to that employed by C. T. R. Wilson. Two parallel horizontal plates were fitted in the vessel and the radiation from an X ray tube or radio-active substance ionized the gas between them. A difference of potential V, small compared with that required to saturate the gas, was applied between the parallel plates distant l cms. from each other. The small current i through the gas is given (section 28) by
where
Since the value of N is the same as the number of drops, and the velocity u is known, the value of e can be determined.
In his last determination J. J. Thomson found that
A very concordant value, namely, 3·1 × 10-10, has been obtained by H. A. Wilson[70], by using a modified method of counting the drops. A check on the size of the drops, determined by their rate of fall, was made by observing the rate at which the drops moved in a strong electric field, arranged so as to act with or against gravity.
J. J. Thomson found that the charge on the ions produced in hydrogen and oxygen is the same. This shows that the nature of the ionization in gases is distinct from that occurring in the electrolysis of solutions where the oxygen ion always carries twice the charge of the hydrogen ion.
37. Diffusion of the ions. Early experiments with ionized gases showed that the conductivity was removed from the gas by passage through a finely divided substance like cotton-wool, or by bubbling through water. This loss of conductivity is due to the fact that the ions in passing through narrow spaces diffuse to the sides of the boundary, to which they either adhere or give up their charge.
A direct determination of the coefficient of diffusion of the ions 52produced in gases by Röntgen rays or by the rays from active substances has been made by Townsend[71]. The general method employed was to pass a stream of ionized gas through a diffusion vessel made up of a number of fine metal tubes arranged in parallel. Some of the ions in their passage through the tubes diffuse to the sides, the proportion being greater the slower the motion of the gas and the narrower the tube. Observations were made of the conductivity of the gas before and after passage through the tubes. In this way, correcting if necessary for the recombination during the time taken to pass through the tubes, the proportion R of either positive or negative ions which are abstracted can be deduced. The value of R can be expressed mathematically by the following equation in terms of K, the coefficient of diffusion of the ions into the gas with which they are mixed[72],
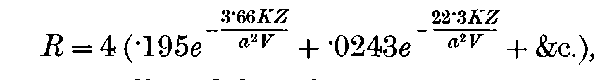
where
Only the first two terms of the series need be taken into account when narrow tubes are used.
In this equation R, V, and a are determined experimentally, and K can thus be deduced.
The following table shows the results obtained by Townsend when X rays were used. Almost identical results were obtained later, when the radiations from active substances replaced the X rays.
| Gas | K for + ions | K for – ions | Mean value of K | Ratio of values of K |
|---|---|---|---|---|
| Air, dry | ·028 | ·043 | ·0347 | 1·54 |
| „ moist | ·032 | ·035 | ·0335 | 1·09 |
| Oxygen, dry | ·025 | ·0396 | ·0323 | 1·58 |
| „ moist | ·0288 | ·0358 | ·0323 | 1·24 |
| Carbonic acid, dry | ·023 | ·026 | ·0245 | 1·13 |
| „ „ moist | ·0245 | ·0255 | ·025 | 1·04 |
| Hydrogen, dry | ·123 | ·190 | ·156 | 1·54 |
| „ moist | ·128 | ·142 | ·135 | 1·11 |
53The moist gases were saturated with water vapour at a temperature of 15° C.
It is seen that the negative ion in all cases diffuses faster than the positive. Theory shows that the coefficients of diffusion should be directly proportional to the velocities of the ions, so that this result is in agreement with the observations on the greater velocity of the negative ion.
This difference in the rate of diffusion of the ions at once explains an interesting experimental result. If ionized gases are blown through a metal tube, the tube gains a negative charge while the gas itself retains a positive charge. The number of positive and negative ions present in the gas is originally the same, but, in consequence of the more rapid diffusion of the negative ions, more of the negative ions than of the positive give up their charges to the tube. The tube consequently gains a negative and the gas a positive charge.
38. A very important result can be deduced at once when the velocities and coefficients of diffusion of the ions are known. Townsend (loc. cit.) has shown that the equation of their motion is expressed by the formula
where e is the charge on an ion,
and u is the velocity due to the electric force X in the direction of the axis of x. When a steady state is reached,
Let N be the number of molecules in a cubic centimetre of gas at the pressure P and at the temperature 15° C., for which the values of u and K have been determined. Then N/P may be substituted for n/p, and, since P at atmospheric pressure is 106,
54then
where u1 is the velocity for 1 volt (i.e. ¹⁄₃₀₀ E. S. unit) per cm.
It is known that one absolute electromagnetic unit of electricity in passing through water liberates 1·23 c.c. of hydrogen at a temperature of 15° C. and standard pressure. The number of atoms in this volume is 2·46N, and, if e´ is the charge on the hydrogen atom in the electrolysis of water,
For example, substituting the values of u1 and K determined in moist air for the positive ion,
Values of this ratio, not very different from unity, are obtained for the positive and negative ions of the gases hydrogen, oxygen, and carbon dioxide. Taking into consideration the uncertainty in the experimental values of u1 and K, these results indicate that the charge carried by an ion in all gases is the same and is equal to that carried by the hydrogen ion in the electrolysis of liquids.
39. Number of the ions. We have seen that, from experimental data, Townsend has found that N, the number of molecules present in 1 c.c. of gas at 15° C. and standard pressure, is given by
Now e, the charge on an ion, is equal to 3·4 × 10-10 E. S. units;
If I is the saturation current through a gas, and q the total rate of production of ions in the gas,
55The saturation current through air was found to be 1·2 × 10-8 ampères, i.e. 36 E.S. units, for parallel plates 4·5 cms. apart, when ·45 gramme of radium of activity 1000 times that of uranium was spread over an area of 33 sq. cms. of the lower plate. This corresponds to a production of about 1011 ions per second. Assuming, for the purpose of illustration, that the ionization was uniform between the plates, the volume of air acted on by the rays was about 148 c.c., and the number of ions produced per c.c. per second about 7 × 108. Since N = 3·6 × 1019, we see that, if one molecule produces two ions, the proportion of the gas ionized per second is about 10-11 of the whole. For uranium the fraction is about 10-14, and for pure radium, of activity one million times that of uranium, about 10-8. Thus even in the case of pure radium, only about one molecule of gas is acted on per second in every 100 millions.
The electrical methods are so delicate that the production of one ion per cubic centimetre per second can be detected readily. This corresponds to the ionization of about one molecule in every 1019 present in the gas.
40. Size and nature of the ions. An approximate estimate of the mass of an ion, compared with the mass of the molecule of the gas in which it is produced, can be made from the known data of the coefficient K of inter-diffusion of the ions into gases. The value of K for the positive ions in moist carbon dioxide has been shown to be ·0245, while the value of K for the inter-diffusion of carbon dioxide with air is ·14. The value of K for different gases is approximately inversely proportional to the square root of the products of the masses of the molecules of the two inter-diffusing gases; thus, the positive ion in carbon dioxide behaves as if its mass were large compared with that of the molecule. Similar results hold for the negative as well as for the positive ion, and for other gases besides carbon dioxide.
This has led to the view that the ion consists of a charged centre surrounded by a cluster of molecules travelling with it, which are kept in position round the charged nucleus by electrical forces. A rough estimate shows that this cluster consists of about 30 molecules of the gas. This idea is supported by the variation in velocity, i.e. the variation of the size of the negative ion, in the 56presence of water vapour; for the negative ion undoubtedly has a greater mass in moist than in dry gases. At the same time it is possible that the apparently large size of the ion, as determined by diffusion methods, may be in part a result of the charge carried by the ion. The presence of a charge on a moving body would increase the frequency of collision with the molecules of the gas, and consequently diminish the rate of diffusion. The ion on this view may not actually be of greater size than the molecule from which it is produced.
The negative and positive ions certainly differ in size, and this difference becomes very pronounced for low pressures of the gas. At atmospheric pressure, the negative ion, produced by the action of ultra-violet light on a negatively charged body, is of the same size as the ion produced by X rays, but at low pressures J. J. Thomson has shown that it is identical with the corpuscle or electron, which has an apparent mass of about ¹⁄₁₀₀₀ of the mass of the hydrogen atom. A similar result has been shown by Townsend to hold for the negative ion produced by X rays at a low pressure. It appears that the negative ion at low pressure sheds its attendant cluster. The result of Langevin, that the velocity of the negative ion increases more rapidly with the diminution of pressure than that of the positive ion, shows that this process of removal of the cluster is quite appreciable at a pressure of 10 mms. of mercury.
We must suppose that the process of ionization in gases consists in a removal of a negative corpuscle or electron from the molecule of the gas. At atmospheric pressure this corpuscle immediately becomes the centre of an aggregation of molecules which moves with it and is the negative ion. After removal of the negative ion the molecule retains a positive charge, and probably also becomes the centre of a cluster of new molecules.
The terms electron and ion as used in this work may therefore be defined as follows:—
The electron or corpuscle is the body of smallest mass yet known to science. It carries a negative charge of value 3·4 × 10-10 electrostatic units. Its presence has only been detected when in rapid motion, when, for speeds up to about 1010 cms. a second, it has an apparent mass m given by e/m = 1·86 × 107 electromagnetic 57units. This apparent mass increases with the speed as the velocity of light is approached (see section 82).
The ions which are produced in gases at ordinary pressure have an apparent size, as determined from their rates of diffusion, large compared with the molecule of the gas in which they are produced. The negative ion consists of an electron with a cluster of molecules attached to and moving with it; the positive ion consists of a molecule from which an electron has been expelled, with a cluster of molecules attached. At low pressures under the action of an electric field the electron does not form a cluster. The positive ion is always atomic in size, even at low pressures of the gas. Each of the ions carries a charge of value 3·4 × 10-10 electrostatic units.
41. Ions produced by collision. The greater part of the radiation from the radio-active bodies consists of a stream of charged particles travelling with great velocity. In this radiation, the α particles, which cause most of the ionization observed in the gas, consist of positively charged bodies projected with a velocity about one-tenth the velocity of light. The β rays consist of negatively charged particles, which are identical with the cathode rays generated in a vacuum tube, and travel with a speed about one-half the velocity of light (chapter IV.). Each of these projected particles, in virtue of its great kinetic energy, sets free a large number of ions by collision with the gas molecules in its path. No definite experimental evidence has yet been obtained of the number of ions produced by a single particle, or of the way in which the ionization varies with the speed, but there is no doubt that each projected body gives rise to many thousand ions in its path before its energy of motion is destroyed.
It has already been mentioned (section 29) that at low pressures ions moving under the action of an electric field are able to produce fresh ions by collision with the molecules of the gas. At low pressures the negative ion is identical with the electron set free in a vacuum tube, or emitted by a radio-active substance.
The mean free path of the ion is inversely proportional to the pressure of the gas. Consequently, if an ion moves in an electric field, the velocity acquired between collisions increases with diminution of the pressure. Townsend has shown that fresh ions are 58occasionally produced by collision when the negative ion moves freely between two points differing in potential by 10 volts. If the difference be about V = 20 volts, fresh ions arise at each collision[73].
Now the energy W, acquired by an ion of charge e moving freely between two points at a difference of potential V, is given by
Taking V = 20 volts = ²⁰⁄₃₀₀ E. S. units, and e = 3·4 × 10-10, the energy W required in the case of a negative ion to produce an ion by collision is given by
The velocity u acquired by the ion of mass m just before a collision is given by
and
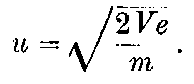
Now e/m = 1·86 × 107 electromagnetic units for the electron at slow speeds (section 82).
Taking V = 20 volts, we find that
This velocity is very great compared with the velocity of agitation of the molecules of the gas.
In a weak electric field, the negative ions only produce ions by collision. The positive ion, whose mass is at least 1000 times greater than the electron, does not acquire a sufficient velocity to generate ions by collision until an electric field is applied nearly sufficient to cause a spark through the gas.
An estimate of the energy required for the production of an ion by X rays has been made by Rutherford and McClung. The energy of the rays was measured by their heating effect, and the total number of ions produced determined. On the assumption that all the energy of the rays is used up in producing ions, it 59was found that V = 175 volts—a value considerably greater than that observed by Townsend from data of ionization by collision. The ionization in the two cases, however, is produced under very different conditions, and it is impossible to estimate how much of the energy of the rays is dissipated in the form of heat.
42. Variations are found in the saturation current through gases, exposed to the radiations from active bodies, when the pressure and nature of the gas and the distance between the electrodes are varied. Some cases which are of special importance in measurements will now be considered. With unscreened active material the ionization of the gas is, to a large extent, due to the α rays, which are absorbed in their passage through a few centimetres of air. In consequence of this rapid absorption, the ionization decreases rapidly from the surface of the active body, and this gives rise to conductivity phenomena different in character from those observed with Röntgen rays, where the ionization is in most cases uniform.
43. Variation of the current with distance between the plates. It has been found experimentally[74] that the intensity of the ionization, due to a large plane surface of active matter, falls off approximately in an exponential law with the distance from the plate. On the assumption that the rate of production of ions at any point is a measure of the intensity I of the radiation, the value of I at that point is given by
where λ is a constant, x the distance from the plate, and I₀ the intensity of the radiation at the surface of the plate.
While the exponential law, in some cases, approximately represents the variation of the ionization with distance, in others the divergence from it is wide. The ionization, due to a plane surface of polonium, for example, falls off more rapidly than the exponential law indicates. The α rays from an active substance like radium are highly complex; the law of variation of the ionization due to them is by no means simple and depends upon a variety of conditions. The distribution of ionization is quite different according as a thick layer or a very thick film of radio-active matter is employed. The question is fully considered at the end of 60chapter IV., but for simplicity, the exponential law is assumed in the following calculations.
Consider two parallel plates placed as in Fig. 1, one of which is covered with a uniform layer of radio-active matter. If the distance d between the plates is small compared with the dimensions of the plates, the ionization near the centre of the plates will be sensibly uniform over any plane parallel to the plates and lying between them. If q be the rate of production of ions at any distance x and q₀ that at the surface, then q = q₀e-λx. The saturation current i per unit area is given by
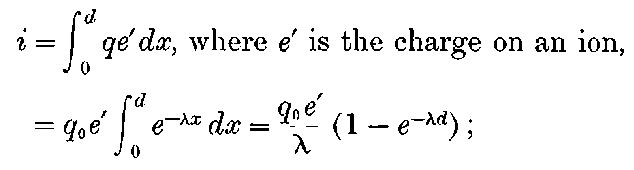
hence, when λd is small, i.e. when the ionization between the plates is nearly constant,
The current is thus proportional to the distance between the plates. When λd is large, the saturation current i₀ is equal to q₀e´/λ, and is independent of further increase in the value of d. In such a case the radiation is completely absorbed in producing ions between the plates, and
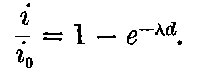
For example, in the case of a thin layer of uranium oxide spread over a large plate, the ionization is mostly produced by rays the intensity of which is reduced to half value in passing through 4·3 mms. of air, i.e. the value of λ is 1·6. The following table is an example of the variation of i with the distance between the plates.
| Distance | Saturation Current |
|---|---|
| 2·5 mms. | 32 |
| 5 „ | 55 |
| 7·5 „ | 72 |
| 10 „ | 85 |
| 12·5 „ | 96 |
| 15 „ | 100 |
Thus the increase of current for equal increments of distance between the plates decreases rapidly with the distance traversed by the radiation.
61The distance of 15 mms. was not sufficient to completely absorb all the radiation, so that the current had not reached its limiting value.
When more than one type of radiation is present, the saturation current between parallel plates is given by
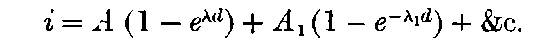
where A, A1 are constants, and λ, λ1 the absorption constants of the radiations in the gas.
Since the radiations are unequally absorbed in different gases, the variation of current with distance depends on the nature of the gas between the plates.
44. Variation of the current with pressure. The rate of production of ions by the radiations from active substances is directly proportional to the pressure of the gas. The absorption of the radiation in the gas also varies directly as the pressure. The latter result necessarily follows if the energy required to produce an ion is independent of the pressure.
In cases where the ionization is uniform between two parallel plates, the current will vary directly as the pressure; when however the ionization is not uniform, on account of the absorption of the radiation in the gas, the current does not decrease directly as the pressure until the pressure is reduced so far that the ionization is sensibly uniform. Consider the variation with pressure of the saturation current i between two large parallel plates, one of which is covered with a uniform layer of active matter.
Let λ1 = absorption constant of the radiation in the gas for unit pressure.
For a pressure p, the intensity I at any point x is given by
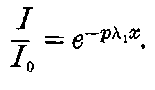
The saturation current i is thus proportional to
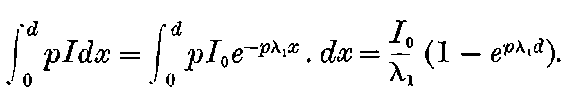
If r be the ratio of the saturation currents for the pressures p1 and p2,
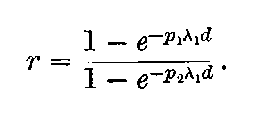
62The ratio is thus dependent on the distance d between the plates and the absorption of the radiation by the gas.
The difference in the shape of the pressure-current curves[75] is well illustrated in Fig. 8, where curves are given for hydrogen, air, and carbonic acid for plates 3·5 cms. apart.
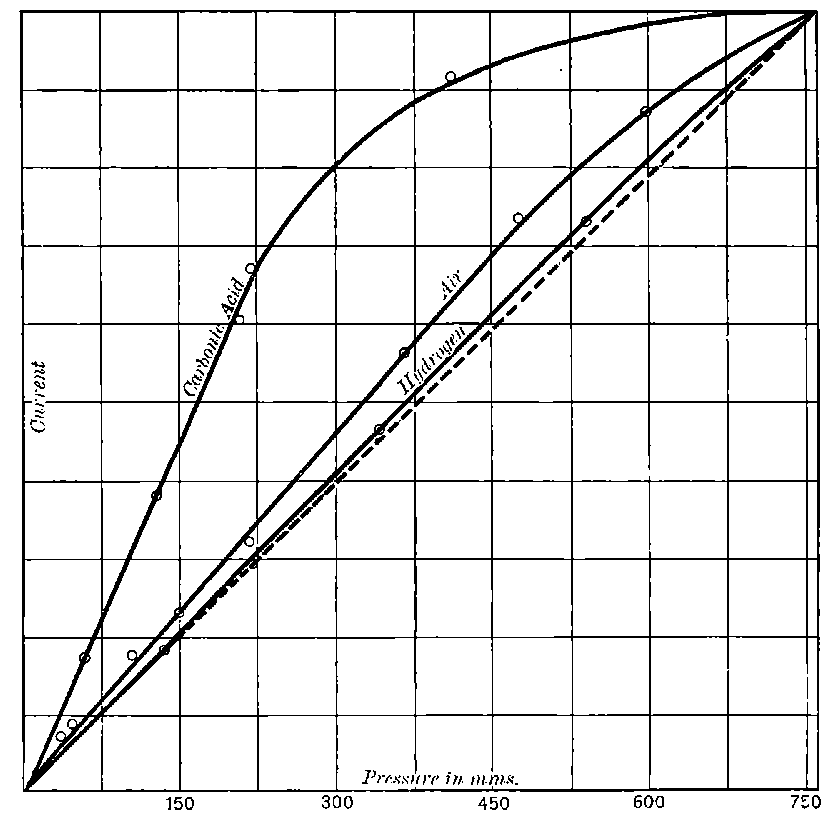
Fig. 8.
For the purpose of comparison, the current at atmospheric pressure and temperature in each case is taken as unity. The actual value of the current was greatest in carbonic acid and least in hydrogen. In hydrogen, where the absorption is small, the current over the whole range is nearly proportional to the pressure. In carbonic acid, where the absorption is large, the current diminishes at first slowly with the pressure, but is nearly proportional to it below the pressure of 235 mms. of mercury. The curve for air occupies an intermediate position.
63In cases where the distance between the plates is large, the saturation current will remain constant with diminution of pressure until the absorption is so reduced that the radiation reaches the other plate.
An interesting result follows from the rapid absorption of radiation by the gas. If the current is observed between two fixed parallel plates, distant d1 and d2 respectively from a large plane surface of active matter, the current at first increases with diminution of pressure, passes through a maximum value, and then diminishes. In such an experimental case the lower plate through which the radiations pass is made either of open gauze or of thin metal foil to allow the radiation to pass through readily.
The saturation current i is obviously proportional to
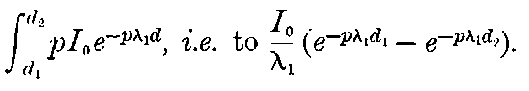
This is a function of the pressure, and is a maximum when
For example, if the active matter is uranium, pλ1 = 1·6 for the α rays at atmospheric pressure. If d2 = 3, and d1 = 1, the saturation current reaches a maximum when the pressure is reduced to about ⅓ of an atmosphere. This result has been verified experimentally.
45. Conductivity of different gases when acted on by the rays. For a given intensity of radiation, the rate of production of ions in a gas varies for different gases and increases with the density of the gas. Strutt[76] has made a very complete examination of the relative conductivity of gases exposed to the different types of rays emitted by active substances. To avoid correction for any difference of absorption of the radiation in the various gases, the pressure of the gas was always reduced until the ionization was directly proportional to the pressure, when, as we have seen above, the ionization must everywhere be uniform throughout the gas. For each type of rays, the ionization of air is taken as unity. The currents through the gases were determined at different pressures, and were reduced to a common 64pressure by assuming that the ionization was proportional to the pressure.
With unscreened active material, the ionization is almost entirely due to α rays. When the active substance is covered with a layer of aluminium ·01 cm. in thickness, the ionization is mainly due to the β or cathodic rays, and when covered with 1 cm. of lead, the ionization is solely due to the γ or very penetrating rays. Experiments on the γ rays of radium were made by observing the rate of discharge of a special gold-leaf electroscope filled with the gas under examination and exposed to the action of the rays. The following table gives the relative conductivities of gases exposed to various kinds of ionizing radiations.
| Gas | Relative Density | α rays | β rays | γ rays | Röntgen rays |
|---|---|---|---|---|---|
| Hydrogen | 0·0693 | 0·226 | 0·157 | 0·169 | 0·114 |
| Air | 1·00 | 1·00 | 1·00 | 1·00 | 1·00 |
| Oxygen | 1·11 | 1·16 | 1·21 | 1·17 | 1·39 |
| Carbon dioxide | 1·53 | 1·54 | 1·57 | 1·53 | 1·60 |
| Cyanogen | 1·86 | 1·94 | 1·86 | 1·71 | 1·05 |
| Sulphur dioxide | 2·19 | 2·04 | 2·31 | 2·13 | 7·97 |
| Chloroform | 4·32 | 4·44 | 4·89 | 4·88 | 31·9 |
| Methyl iodide | 5·05 | 3·51 | 5·18 | 4·80 | 72·0 |
| Carbon tetrachloride | 5·31 | 5·34 | 5·83 | 5·67 | 45·3 |
With the exception of hydrogen, it will be seen that the ionization of gases is approximately proportional to their density for the α, β, γ rays of radium. The results obtained by Strutt for Röntgen rays are quite different; for example, the relative conductivity produced by them in methyl iodide was more than 14 times as great as that due to the rays of radium. The relative conductivities of gases exposed to X rays has been recently re-examined by McClung[77] and Eve[78], who have found that the conductivity depends upon the penetrating power of the X rays employed. The results obtained by them will be discussed later (section 107).
65This difference of conductivity in gases is due to unequal absorptions of the radiations. The writer has shown[79] that the total number of ions produced by the α rays for uranium, when completely absorbed by different gases, is not very different. The following results were obtained:
| Gas | Total Ionization |
| Air | 100 |
| Hydrogen | 95 |
| Oxygen | 106 |
| Carbonic acid | 96 |
| Hydrochloric acid gas | 102 |
| Ammonia | 101 |
The numbers, though only approximate in character, seem to show that the energy required to produce an ion is probably not very different for the various gases. Assuming that the energy required to produce an ion in different gases is about the same, it follows that the relative conductivities are proportional to the relative absorption of the radiations.
A similar result has been found by McLennan for cathode rays. He proved that the ionization was directly proportional to the absorption of the rays in the gas, thus showing that the same energy is required to produce an ion in all the gases examined.
46. Potential Gradient. The normal potential gradient between two charged electrodes is always disturbed when the gas is ionized in the space between them. If the gas is uniformly ionized between two parallel plates, Child and Zeleny have shown that there is a sudden drop of potential near the surface of both plates, and that the electric field is sensibly uniform for the intermediate space between them. The disturbance of the potential gradient depends upon the difference of potential applied, and is different at the surface of the two plates.
In most measurements of radio-activity the material is spread over one plate only. In such a case the ionization is to a large extent confined to the volume of the air close to the active plate. The potential gradient in such a case is shown in Fig. 9. The 66dotted line shows the variation of potential at any point between the plates when no ionization is produced between the plates; curve A for weak ionization, such as is produced by uranium, curve B for the intense ionization produced by a very active substance. In both cases the potential gradient is least near the active plate, and greatest near the opposite plate. For very intense ionization it is very small near the active surface. The potential gradient varies slightly according as the active plate is charged positively or negatively.
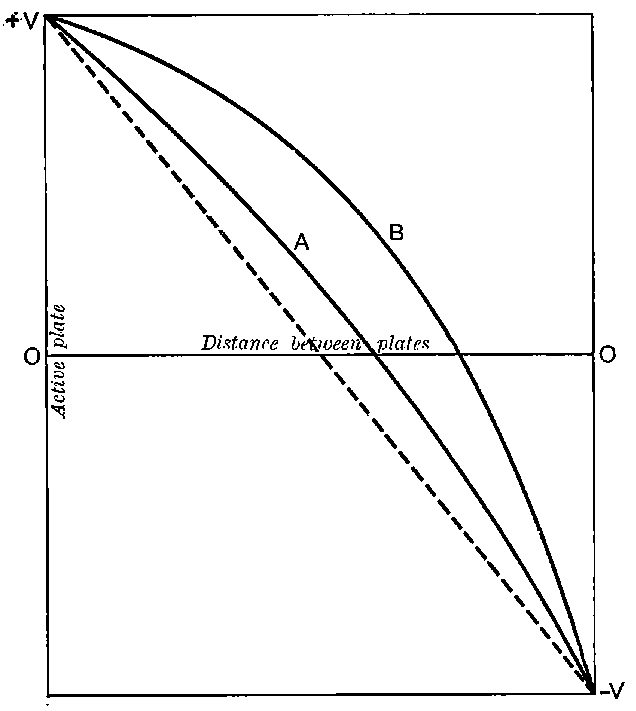
Fig. 9.
47. Variation of current with voltage for surface ionization.
Some very interesting results, giving the variation of the current with voltage, are observed when the ionization is intense, and confined to the space near the surface of one of two parallel plates between which the current is measured.
The theory of this subject has been worked out independently by Child[80] and Rutherford[81]. Let V be the potential difference 67between two parallel plates at a distance d apart. Suppose that the ionization is confined to a thin layer near the surface of the plate A (see Fig. 1) which is charged positively. When the electric field is acting, there is a distribution of positive ions between the plates A and B.
The current i1 per square centimetre through the gas is constant for all values of x, and is given by
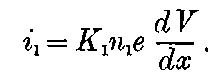
By Poisson’s equation
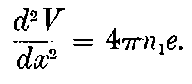
Then
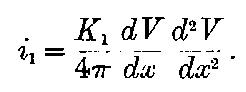
Integrating
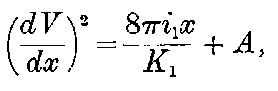
where A is a constant. Now A is equal to the value of
when x = 0. By making the ionization very intense, the value of
can be made extremely small.
Putting A = 0, we see that
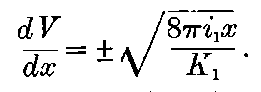
This gives the potential gradient between the plates for different values of x.
Integrating between the limits 0 and d,
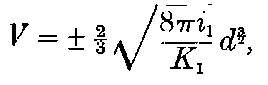
or
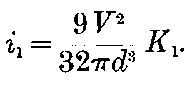
68If i2 is the value of the current when the electric field is reversed, and K2 the velocity of the negative ion,
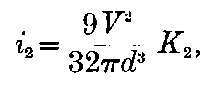
and
The current in the two directions is thus directly proportional to the velocities of the positive and negative ions. The current should vary directly as the square of the potential difference applied, and inversely as the cube of the distance between the plates.
The theoretical condition of surface ionization cannot be fulfilled by the ionization due to active substances, as the ionization extends some centimetres from the active plate. If, however, the distance between the plates is large compared with the distance over which the ionization extends, the results will be in rough agreement with the theory. Using an active preparation of radium, the writer has made some experiments on the variation of current with voltage between parallel plates distant about 10 cms. from each other[82].
The results showed
(1) That the current through the gas for small voltages increased more rapidly than the potential difference applied, but not as rapidly as the square of that potential difference.
(2) The current through the gas depended on the direction of the electric field; the current was always smaller when the active plate was charged positively on account of the smaller mobility of the positive ion. The difference between i1 and i2 was greatest when the gas was dry, which is the condition for the greatest difference between the velocities of the ions.
An interesting result follows from the above theory. For given values of V and d, the current cannot exceed a certain definite value, however much the ionization may be increased. In a similar way, when an active preparation of radium is used as a source of surface ionization, it is found that, for a given voltage 69and distance between the plates, the current does not increase beyond a certain value however much the activity of the material is increased.
48. Magnetic field produced by an ion in motion. It will be shown later that the two most important kinds of rays emitted by radio-active substances consist of electrified particles, spontaneously projected with great velocity. The easily absorbed rays, known as α rays, are positively electrified atoms of matter; the penetrating rays, known as β rays, carry a negative charge, and have been found to be identical with the cathode rays produced by the electric discharge in a vacuum tube.
The methods adopted to determine the character of these rays are very similar to those first used by J. J. Thomson to show that the cathode rays consisted of a stream of negatively electrified particles projected with great velocity.
The proof that the cathode rays were corpuscular in character, and consisted of charged particles whose mass was very small compared with that of the hydrogen atom, marked an important epoch in physical science: for it not only opened up new and fertile fields of research, but also profoundly modified our previous conceptions of the constitution of matter.
A brief account will accordingly be given of the effects produced by a moving charged body, and also of some of the experimental methods which have been used to determine the mass and velocity of the particles of the cathode stream[83].
Consider an ion of radius a, carrying a charge of electricity e, and moving with a velocity u, small compared with the velocity of light. In consequence of the motion, a magnetic field is set up around the charged ion, which is carried with it. The charged ion in motion constitutes a current element of magnitude eu, and the magnetic field H at any point distant r from the sphere is given by
70where θ is the angle the radius vector makes with the direction of motion. The lines of magnetic force are circles around the axis of motion. When the ion is moving with a velocity small compared with the velocity of light, the lines of electric force are nearly radial, but as the speed of light is approached, they tend to leave the axis of motion and to bend towards the equator. When the speed of the body is very close to that of light, the magnetic and electric field is concentrated to a large extent in the equatorial plane.
The presence of a magnetic field around the moving body implies that magnetic energy is stored up in the medium surrounding it. The amount of this energy can be calculated very simply for slow speeds.
In a magnetic field of strength H, the magnetic energy stored up in unit volume of the medium of unit permeability is given by
Integrating the value of this expression over the region exterior to a sphere of radius a, the total magnetic energy due to the motion of the charged body is given by
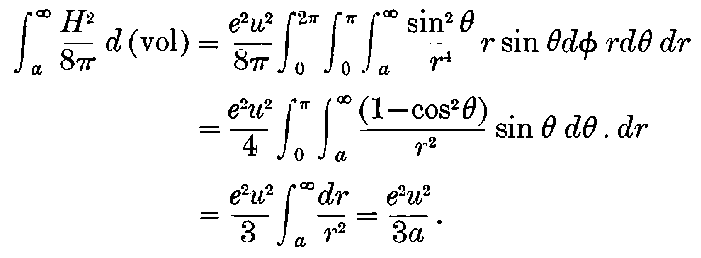
The magnetic energy, due to the motion, is analogous to kinetic energy, for it depends upon the square of the velocity of the body. In consequence of the charge carried by the ion, additional kinetic energy is associated with it. If the velocity of the ion is changed, electric and magnetic forces are set up tending to stop the change of motion, and more work is done during the change than if the ion were uncharged. The ordinary kinetic energy of the body is
In consequence of its charge, the kinetic energy associated with it is increased by
It thus behaves as if it possessed a mass m + m1 where m1 is the electrical mass, with the value
71We have so far only considered the electrical mass of a charged ion moving with a velocity small compared with that of light. As the speed of light is approached, the magnetic energy can no longer be expressed by the equation already given. The general values of the electrical mass of a charged body for speed were first worked out by J. J. Thomson[84] in 1887. A more complete examination was made in 1889 by Heaviside[85], while Searle[86] worked out the case for a charged ellipsoid. Recently, the question was again attacked by Abraham[87]. Slightly different expressions for the variation of electrical mass with speed have been obtained, depending upon the conditions assumed for the distribution of the electricity on the sphere. The expression found by Abraham, which has been utilized by Kaufmann to show that the mass of the electron is electromagnetic in origin, is given later in section 82.
All the calculations agree in showing that the electrical mass is practically constant for slow speeds, but increases as the speed of light is approached, and is theoretically infinite when the speed of light is reached. The nearer the velocity of light is approached, the greater is the resisting force to a change of motion. An infinite force would be required to make an electron actually attain the velocity of light, so that, according to the present theory, it would be impossible for an electron to move faster than light, i.e. faster than an electromagnetic disturbance travels in the ether.
The importance of these deductions lies in the fact that an electric charge in motion, quite independently of any material nucleus, possesses an apparent mass in virtue of its motion, and that this mass is a function of the speed. Indeed, we shall see later (see section 82) that the apparent mass of the particles constituting the cathode stream can be explained in virtue of their charge, without the necessity of assuming a material body in which the charge is distributed. This has led to the suggestion that all mass may be electrical in origin, and due purely to electricity in motion.
49. Action of a magnetic field on a moving ion. Let us consider the case of an ion of mass m carrying a charge e and 72moving freely with a velocity u. If u is small compared with the velocity of light, the ion in motion corresponds to a current element of magnitude eu. If the ion moves in an external magnetic field of strength H, it is acted on by a force at right angles both to the direction of motion, and to that of the magnetic force and equal in magnitude to Heu sin θ, where θ is the angle between the direction of the magnetic force and the direction of motion. Since the force due to the magnetic field is always perpendicular to the direction of motion, it has no effect upon the velocity of the particle, but can only alter the direction of its path.
If ρ is the radius of curvature of the path of the ion, the force along the normal is equal to
and this is balanced by the force Heu sin θ.
If
i.e. if the ion is moving at right angles to the direction of the magnetic field
or
Since u is constant, ρ is also constant, i.e. the particle describes a circular orbit of radius ρ. The radius of the circular orbit is thus directly proportional to u, and inversely proportional to H.
If the ion is moving at an angle θ with the direction of the magnetic field, it describes a curve which is compounded of a motion of a particle of velocity u sin θ perpendicular to the field and u cos θ in the direction of the field. The former describes a circular orbit of radius ρ, given by
the latter is unaffected by the magnetic field and moves uniformly in the direction of the magnetic field with a velocity u cos θ. The motion of the particle is in consequence a helix, traced on a cylinder of radius
whose axis is in the direction of the magnetic field. Thus an ion projected obliquely to the direction of a uniform magnetic field always moves in a helix whose axis is parallel to the lines of magnetic force[88].
73 50. Determination of e/m for the cathode stream. The cathode rays, first observed by Varley, were investigated in detail by Crookes. These rays are projected from the cathode in a vacuum tube at low pressure. They travel in straight lines, and are readily deflected by a magnet, and produce strong luminosity in a variety of substances placed in their path. The rays are deflected by a magnetic field in the same direction as would be expected for a negatively charged particle projected from the cathode. In order to explain the peculiar properties of these rays Crookes supposed that they consisted of negatively electrified particles, moving with great velocity and constituting, as he appropriately termed it, “a new or fourth state of matter.” The nature of these rays was for twenty years a subject of much controversy, for while some upheld their material character, others considered that they were a special form of wave motion in the ether.
Perrin and J. J. Thomson showed that the rays always carried with them a negative charge, while Lenard made the important discovery that the rays passed through thin metal foil and other substances opaque to ordinary light. Using this property, he sent the rays through a thin window and examined the properties of the rays outside the vacuum tube in which they were produced.
The absorption of the rays by matter was shown to be nearly proportional to the density over a very wide range, and to be independent of its chemical constitution.
The nature of these rays was successfully demonstrated by J. J. Thomson[89] in 1897. If the rays consisted of negatively electrified particles, they should be deflected in their passage through an electric as well as through a magnetic field. Such an experiment had been tried by Hertz, but with negative results. J. J. Thomson, however, found that the rays were deflected by an electric field in the direction to be expected for a negatively charged particle, and showed that the failure of Hertz to detect the same was due to the masking of the electric field by the strong ionization produced in the gas by the cathode stream. This effect was got rid of by reducing the pressure of the gas in the tube.
74The experimental arrangement used for the electric deflection of the rays is shown in Fig. 10.
The cathode rays are generated at the cathode C, and a narrow pencil of rays is obtained by passing the rays through a perforated disc AB. The rays then passed midway between two parallel insulated plates D and E, d centimetres apart, and maintained at a constant difference of potential V. The point of incidence of the pencil of rays was marked by a luminous patch produced on a phosphorescent screen placed at PP´.
The particle carrying a negative charge e in passing between the charged plates, is acted on by a force Xe directed towards the positive plate, where X, the strength of the electric field, is given by
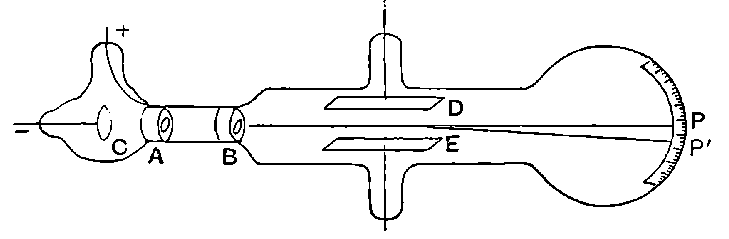
Fig. 10.
The application of the electric field thus causes the luminous patch to move in the direction of the positive plate. If now a uniform magnetic field is applied at the plates D and E, perpendicular to the pencil of rays, and parallel to the plane of the plates, and in such a direction that the electric and magnetic forces are opposed to one another, the patch of light can be brought back to its undisturbed position by adjusting the strength of the magnetic field. If H is the strength of the magnetic field, the force on the particle due to the magnetic field is Heu, and when a balance is obtained
Now if the magnetic field H is acting alone, the curvature ρ of the path of the rays between the plates can be deduced from the deflection of the luminous patch. But we have seen that
75From equations (1) and (2), the value of u and e/m for the particle can be determined.
The velocity u is not constant, but depends upon the potential difference between the electrodes, and this in turn depends upon the pressure and nature of the residual gas in the tube.
By altering these factors, the cathode particles may be made to acquire velocities varying between about 109 and 1010 cms. per second. This velocity is enormous compared with that which can be impressed ordinarily upon matter by mechanical means. On the other hand, the value of e/m for the particles is sensibly constant for different velocities.
As a result of a series of experiments the mean value e/m = 7·7 × 106 was obtained. The value of e/m is independent of the nature or pressure of the gas in the vacuum tube and independent of the metal used as cathode. A similar value of e/m was obtained by Lenard[90] and others.
Kaufmann[91] and Simon[92] used a different method to determine the value of e/m. The potential difference V between the terminals of the tube was measured. The work done on the charged particle in moving from one end of the tube to the other is Ve, and this must be equal to the kinetic energy
acquired by the moving particle. Thus
By combination of this equation with (2) obtained by measurement of the magnetic deflexion, both u and e/m can be determined.
Simon found by this method that
76It will be seen later (section 82) that a similar value was deduced by Kaufmann for the electrons projected from radium.
These results, which have been based on the effect of a magnetic and electric field on a moving ion, were confirmed by Weichert, who determined by a direct method the time required for the particle to traverse a known distance.
The particles which make up the cathode stream were termed “corpuscles” by J. J. Thomson. The name “electron,” first employed by Johnstone Stoney, has also been applied to them and has come into general use[93].
The methods above described do not give the mass of the electron, but only the ratio of the charge to the mass. A direct comparison can, however, be made between the ratio e/m for the electron and the corresponding value for the hydrogen atoms set free in the electrolysis of water. Each of the hydrogen atoms is supposed to carry a charge e, and it is known that 96,000 coulombs of electricity, or, in round numbers, 104 electromagnetic units of quantity are required to liberate one gram of hydrogen. If N is the number of atoms in one gram of hydrogen, then Ne = 104. But if m is the mass of a hydrogen atom, then Nm = 1. Dividing one by the other e/m = 104. We have seen already that a gaseous ion carries the same charge as a hydrogen atom, while indirect evidence shows that the electron carries the same charge as an ion, and consequently the same charge as the atom of hydrogen. Hence we may conclude that the apparent mass of the electron is only about ¹⁄₁₀₀₀ of the mass of the hydrogen atom. The electron thus behaves as the smallest body known to science.
In later experiments J. J. Thomson showed that the negative ions set free at low pressures by an incandescent carbon filament, and also the negative ions liberated from a zinc plate exposed to the action of ultra-violet light, had the same value for e/m as the 77electrons produced in a vacuum tube. It thus seemed probable that the electron was a constituent of all matter. This view received strong support from measurements of quite a different character. Zeeman in 1897 found that the lines of the spectrum from a source of light exposed in a strong magnetic field were displaced and doubled. Later work has shown that the lines in some cases are trebled, in others sextupled, while, in a few cases, the multiplication is still greater. These results received a general explanation on the radiation theories previously advanced by Lorenz and Larmor. The radiation, emitted from any source, was supposed to result from the orbital or oscillatory motion of the charged parts constituting the atom. Since a moving ion is acted on by an external magnetic field, the motion of the charged ions is disturbed when the source of light is exposed between the poles of a strong magnet. There results a small change in the period of the emitted light, and a bright line in the spectrum is, in consequence, displaced by the action of the magnetic field. According to theory, the small change in the wave-length of the emitted light depends upon the strength of the magnetic field and on the ratio e/m of the charge carried by the ion to its mass. By comparison of the theory with the experimental results, it was deduced that the moving ion carried a negative charge, and that the value of e/m was about 107. The charged ion, responsible for the radiation from a luminous body, is thus identical with the electron set free in a vacuum tube.
It thus seems reasonable to suppose that the atoms of all bodies are complex and are built up, in part at least, of electrons, whose apparent mass is very small compared with that of the hydrogen atom. The properties of such disembodied charges has been examined mathematically among others by Larmor, who sees in this conception the ultimate basis of a theory of matter. J. J. Thomson and Lord Kelvin have investigated mathematically certain arrangements of a number of electrons which are stable for small disturbances. This question will be discussed more in detail in section 270.
78 51. Canal rays. If a discharge is passed through a vacuum tube provided with a perforated cathode, within certain limits of pressure, luminous streams are observed to pass through the holes and to emerge on the side of the cathode remote from the anode. These rays were first observed by Goldstein[94] and were called by him the “Canal-strahlen.” These rays travel in straight lines and produce phosphorescence in various substances.
Wien[95] showed that the canal rays were deflected by strong magnetic and electric fields, but the amount of deflection was very small compared with that of the cathode rays under similar conditions. The deflection was found to be opposite in direction to the cathode rays, and this indicates that the canal rays consist of positive ions. Wien determined their velocity and the ratio e/m, by measuring the amount of their magnetic and electric deflection. The value of e/m was found to be variable, depending upon the gas in the tube, but the maximum value observed was 104. This shows that the positive ion, in no case, has a mass less than that of the hydrogen atom. It seems probable that the canal rays consist of positive ions, derived either from the gas or the electrodes, which travel towards the cathode, and have sufficient velocity to pass through the holes of the cathode and to appear in the gas beyond.
It is remarkable that, so far, no case has been observed where the carrier of a positive charge has an apparent mass less than that of the hydrogen atom. Positive electricity always appears to be associated with bodies atomic in size. We have seen that the process of ionization in gases is supposed to consist of the expulsion of an electron from the atom. The corresponding positive charge remains behind on the atom and travels with it. This difference between positive and negative electricity appears to be fundamental, and no explanation of it has, as yet, been forthcoming.
52. Radiation of energy. If an electron moves uniformly in a straight line with constant velocity, the magnetic field, which 79travels with it, remains constant, and there is no loss of energy from it by radiation. If, however, its motion is hastened or retarded, the magnetic field is altered, and there results a loss of energy from the electron in the form of electromagnetic radiation. The rate of loss of energy from an accelerated electron was first calculated by Larmor[96] and shown to be
where e is the charge on the electron in electromagnetic units, and V the velocity of light.
Any alteration in the velocity of a moving charge is thus always accompanied by a radiation of energy from it. Since the electron, set free in a vacuum tube, increases in velocity in passing through the electric field, energy must be radiated from it during its passage from cathode to anode. It can, however, readily be calculated that, in ordinary cases, this loss of energy is small compared with the kinetic energy acquired by the electron in passing through the electric field.
An electron moving in a circular orbit is a powerful radiator of energy, since it is constantly accelerated towards the centre. An electron moving in an orbit of radius equal to the radius of an atom (about 10-8 cms.) would lose most of its kinetic energy of motion in a small fraction of a second, even though its velocity was originally nearly equal to the velocity of light. If, however, a number of electrons are arranged at equal angular intervals on the circumference of a circle and move with constant velocity round the ring, the radiation of energy is much less than for a single electron, and rapidly diminishes with an increase in the number of electrons round the ring. This result, obtained by J. J. Thomson, will be discussed in more detail later when the stability of systems composed of rotating electrons is under consideration.
Since the radiation of energy is proportional to the square of the acceleration, the proportion of the total energy radiated depends upon the suddenness with which an electron is started or stopped. Now some of the cathode ray particles are stopped abruptly when they impinge on the metal cathode, and, in consequence, give up a fraction of their kinetic energy in the form of electromagnetic radiation. Stokes and Weichert suggested that this radiation 80constituted the X rays, which are known to have their origin at the surface on which the cathode rays impinge. The mathematical theory has been worked out by J. J. Thomson[97]. If the motion of an electron is suddenly arrested, a thin spherical pulse in which the magnetic and electric forces are very intense travels out from the point of impact with the velocity of light. The more suddenly the electron is stopped, the thinner and more intense is the pulse. On this view the X rays are not corpuscular like the cathode rays, which produce them, but consist of transverse disturbances in the ether, akin in some respects to light waves of short wave-length. The rays are thus made up of a number of pulses, which are non-periodic in character, and which follow one another at irregular intervals.
On this theory of the nature of the X rays, the absence of direct deflection, refraction, or polarization is to be expected, if the thickness of the pulse is small compared with the diameter of an atom. It also explains the non-deflection of the path of the rays by a magnetic or electric field. The intensity of the electric and magnetic force in the pulse is so great that it is able to cause a removal of an electron from some of the atoms of the gas, over which the pulse passes, and thus causes the ionization observed.
The cathode rays produce X rays, and these in turn give rise to a secondary radiation whenever they impinge on a solid body. This secondary radiation is emitted equally in all directions, and consists partly of a radiation of the X ray type and also of electrons projected with considerable velocity. This secondary radiation gives rise to a tertiary radiation and so on.
Barkla[98] has shown that the secondary radiation emitted from a gas through which the rays pass consists in part of scattered X rays of about the same penetrating power as the primary rays as well as some easily absorbed rays.
Part of the cathode rays is diffusely reflected on striking the cathode. These scattered rays consist in part of electrons of the same speed as in the primary beam, but also include some others of much less velocity. The amount of diffuse reflection depends upon the nature of the cathode and the angle of incidence of the rays.
81We shall see later (chapter IV.) that similar effects are produced when the rays from radio-active substances impinge upon solid bodies.
In this chapter an account of the ionization theory of gases has been given to the extent that is necessary for the interpretation of the measurements of radio-activity by the electric method. It would be out of place here to discuss the development of that theory in detail, to explain the passage of electricity through flames and vapours, the discharge of electricity from hot bodies, and the very complicated phenomena observed in the passage of electricity through a vacuum tube.
For further information on this important subject, the reader is referred to J. J. Thomson’s Conduction of Electricity through Gases, in which the whole subject is treated in a full and complete manner. A simple account of the effect of moving charges and the electronic theory of matter was given by the same author in the Silliman Lectures of Yale University and published under the title Electricity and Matter (Scribner, New York, 1904).
53. Methods of Measurement. Three general methods have been employed for examination of the radiations from radio-active bodies, depending on
The third method is very restricted in its application, and can only be employed for intensely active substances like radium or polonium.
The photographic method has been used very widely, especially in the earlier development of the subject, but has gradually been displaced by the electrical method, as a quantitative determination of the radiations became more and more necessary. In certain directions, however, it possesses distinct advantages over the electrical method. For example, it has proved a very valuable means of investigating the curvature of the path of the rays, when deflected by a magnetic or electric field, and has allowed us to determine the constants of these rays with considerable accuracy.
On the other hand, as a general method of study of the radiations, it is open to many objections. A day’s exposure is generally required to produce an appreciable darkening of the sensitive film when exposed to a weak source of radiation like uranium and thorium. It cannot, in consequence, be employed to investigate the radiations of those active products which 83rapidly lose their activity. Moreover, W. J. Russell has shown that the darkening of a photographic plate can be produced by many agents which do not give out rays like those of the radio-active bodies. This darkening of the plate is produced under the most varied conditions, and very special precautions are necessary when long exposures to a weak source of radiation are required.
The main objection to the photographic method, however, lies in the fact that the radiations which produce the strongest electrical effect are very weak photographically. For example, Soddy[99] has shown that the photographic action of uranium is due almost entirely to the more penetrating rays, and that the easily absorbed rays produce in comparison very little effect. Speaking generally, the penetrating rays are the most active photographically, and, under ordinary conditions, the action on the plate is almost entirely due to them.
Most of the energy radiated from active bodies is in the form of easily absorbed rays which are comparatively inactive photographically. These rays are difficult to study by the photographic method, as the layer of black paper which, in many cases, is required in order to absorb the phosphorescent light from active substances, cuts off at the same time most of the rays under examination. These easily absorbed rays will be shown to play a far more important part in the processes occurring in radio-active bodies than the penetrating rays which are more active photographically.
The electrical method, on the other hand, offers a rapid and accurate method of quantitatively examining the radiations. It can be used as a means of measurement of all the types of radiation emitted, excluding light waves, and is capable of accurate measurement over an extremely wide range. With proper precautions it can be used to measure effects produced by radiations of extremely small intensity.
54. Electrical Methods. The electrical methods employed in studying radio-activity are all based on the property of the radiation in question of ionizing the gas, i.e. of producing positively and negatively charged carriers throughout the volume of the gas. The discussion of the application of the ionization theory of gases to 84measurements of radio-activity has been given in the last chapter. It has been shown there that the essential condition to be fulfilled for comparative measurements of the intensity of the radiations is that the electrical field shall in all cases be strong enough to obtain the maximum or saturation current through the gas.
The electric field required to produce practical saturation varies with the intensity of the ionization and consequently with the activity of the preparations to be examined. For preparations which have an activity not more than 500 times that of uranium, under ordinary conditions, a field of 100 volts per cm. is sufficient to produce a practical saturation current. For very active samples of radium, it is often impossible to obtain conveniently a high enough electromotive force to give even approximate saturation. Under such conditions comparative measurement can be made by measuring the current under diminished pressure of the gas, when saturation is more readily obtained.
The method to be employed in the measurement of this ionization current depends largely on the intensity of the current to be measured. If some very active radium is spread on the lower of two insulated plates as in Fig. 1, and a saturating electric field applied, the current may readily be measured by a sensitive galvanometer of high resistance. For example, a weight of ·45 gr. of radium chloride of activity 1000 times that of uranium oxide, spread over a plate of area 33 sq. cms., gave a maximum current of 1·1 × 10-8 amperes when the plates were 4·5 cms. apart. In this case the difference of potential to be applied to produce practical saturation was about 600 volts. Since most of the ionization is due to rays which are absorbed in passing through a few centimetres of air, the current is not much increased by widening the distance between the two plates. In cases where the current is not quite large enough for direct deflection, the current may be determined by connecting the upper insulated plate with a well insulated condenser. After charging for a definite time, say one or more minutes, the condenser is discharged through the galvanometer, and the current can readily be deduced.
55. In most cases, however, when dealing with less active substances like uranium or thorium, or with small amounts of active 85material, it is necessary to employ methods for measuring much smaller currents than can be detected conveniently by an ordinary galvanometer. The most convenient apparatus to employ for this purpose is one of the numerous types of quadrant electrometer or an electroscope of special design. For many observations, especially where the activity of the two substances is to be compared under constant conditions, an electroscope offers a very certain and easy method of measurement. As an example of a simple apparatus of this kind, a brief description will be given of the electroscope used by M. and Mme Curie in many of their earlier observations.
Fig. 11.
The connections are clearly seen from Fig. 11. The active material is placed on a plate laid on top of the fixed circular plate P, connected with the case of the instrument and with earth. The upper insulated plate P´ is connected with the insulated gold-leaf system LL´. S is an insulating support and L the gold-leaf.
The system is first charged to a suitable potential by means of the rod C. The rate of movement of the gold-leaf is observed by means of a microscope. In comparisons of the activity of two specimens, the time taken by the gold leaf to pass over a certain number of divisions of the micrometer scale in the eye-piece is observed. Since the capacity of the charged system is constant, the average rate of movement of the gold-leaf is directly proportional to the ionization current between P and P´, i.e. to the intensity of the radiation emitted by the active substance. Unless very active 86material is being examined, the difference of potential between P and P´ can easily be made sufficient to produce saturation.
When necessary, a correction can be made for the rate of leak when no active material is present. In order to avoid external disturbances, the plates PP´ and the rod C are surrounded by metal cylinders, E and F, connected with earth.
56. A modified form of the gold-leaf electroscope can be used to determine extraordinarily minute currents with accuracy, and can be employed in cases where a sensitive electrometer is unable to detect the current. A special type of electroscope has been used by Elster and Geitel, in their experiments on the natural ionization of the atmosphere. A very convenient type of electroscope to measure the current due to minute ionization of the gas is shown in Fig. 12.
Fig. 12.
This type of instrument was first used by C. T. R. Wilson[100] in his experiments of the natural ionization of air in closed vessels. A brass cylindrical vessel is taken of about 1 litre capacity. The gold-leaf system, consisting of a narrow strip of gold-leaf L attached to a flat rod R, is insulated inside the vessel by the small sulphur bead or piece of amber S, supported from the rod P. In a dry atmosphere a clean sulphur bead or piece of amber is almost a perfect insulator. The system is charged by a light bent rod CC´ passing through an ebonite cork[101]. The rod C is connected to one terminal of a battery of small accumulators of 200 to 300 volts. If these are absent, the system can be charged by means of a rod of sealing-wax. The charging rod CC´ is then removed from contact with the gold-leaf system. The rods P and C and the cylinder are then connected with earth.
The rate of movement of the gold-leaf is observed by a reading 87microscope through two holes in the cylinder, covered with thin mica. In cases where the natural ionization due to the enclosed air in the cylinder is to be measured accurately, it is advisable to enclose the supporting and charging rod and sulphur bead inside a small metal cylinder M connected to earth, so that only the charged gold-leaf system is exposed in the main volume of the air.
In an apparatus of this kind the small leakage over the sulphur bead can be eliminated almost completely by keeping the rod P charged to the average potential of the gold-leaf system during the observation. This method has been used with great success by C. T. R. Wilson (loc. cit.). Such refinements, however, are generally unnecessary, except in investigations of the natural ionization of gases at low pressures, when the conduction leak over the sulphur bead is comparable with the discharge due to the ionized gas.
57. The electric capacity C of a gold-leaf system about 4 cms. long is usually about 1 electrostatic unit. If V is the decrease of potential of the gold-leaf system in t seconds, the current i through the gas is given by
With a well cleaned brass electroscope of volume 1 litre, the fall of potential due to the natural ionization of the air was found to be about 6 volts per hour. Since the capacity of the gold-leaf system was about 1 electrostatic unit
With special precautions a rate of discharge of ⅒ or even ¹⁄₁₀₀ of this amount can be measured accurately.
The number of ions produced in the gas can be calculated if the charge on an ion is known. J. J. Thomson has shown that the charge e on an ion is equal to 3·4 × 10-10 electrostatic units or 1·13 × 10-19 coulombs.
If the ionization be uniform, the saturation current i is given by i = qSe.
88Now for an electroscope with a volume of 1000 c.c., i was equal to about 1·9 × 10-15 amperes. Substituting the values given above
With suitable precautions an electroscope can thus readily measure an ionization current corresponding to the production of 1 ion per cubic centimetre per second.
The great advantage of an apparatus of this kind lies in the fact that the current measured is due to the ionization inside the vessel and is not influenced by the ionization of the external air or by electrostatic disturbances[102]. Such an apparatus is very convenient for investigating the very penetrating radiations from the radio-elements, since these rays pass readily through the walls of the electroscope. When the electroscope is placed on a lead plate 3 or 4 mms. thick, the ionization in the electroscope, due to a radio-active body placed under the lead, is due entirely to the very penetrating rays, since the other two types of rays are completely absorbed in the lead plate. If a circular opening is cut in the base of the electroscope and covered with thin aluminium of sufficient thickness to absorb the α rays, measurements of the intensity of the β rays from an active substance placed under it, can be made with ease and certainty.
58. A modified form of electroscope, which promises to be of great utility for measuring currents even more minute than those to be observed with the type of instrument already described, has recently been devised by C. T. R. Wilson[103]. The construction of the apparatus is shown in Fig. 13.
The case consists of a rectangular brass box 4 cms. × 4 cms. × 3 cms. A narrow gold-leaf L is attached to a rod R passing through a clean sulphur cork. Opposite the gold-leaf is fixed an insulated brass plate P, placed about 1 mm. from the wall of the box. The movement of the gold-leaf is observed through two small windows by means of a microscope provided with a micrometer scale. The plate P is maintained at a constant potential (generally 89about 200 volts). The electrometer case is placed in an inclined position as shown in the figure, the angle of inclination and the potential of the plate being adjusted to give the desired sensitiveness. The gold-leaf is initially connected to the case, and the microscope adjusted so that the gold-leaf is seen in the centre of the scale. For a given potential of the plate, the sensitiveness depends on the angle of tilt of the case. There is a certain critical inclination below which the gold-leaf is unstable. The most sensitive position lies just above the critical angle. In a particular experiment Wilson found that with an angle of tilt of 30° and with the plate at a constant potential of 207 volts, the gold-leaf, when raised to a potential of one volt above the case, moved over 200 scale divisions of the eye-piece, 54 divisions corresponding to one millimetre.
Fig. 13.
In use, the rod R is connected with the external insulated system whose rise or fall of potential is to be measured. On account of the small capacity of the system and the large movement of the gold-leaf for a small difference of potential, the electroscope is able to measure extraordinarily minute currents. The apparatus is portable. If the plate P be connected to one pole of a dry pile the gold-leaf is stretched out towards the plate, and in this position can be carried without risk of injury.
9059. Electrometers. Although the electroscope can be used with advantage in special cases, it is limited in its application. The most generally convenient apparatus for measurement of ionization currents through gases is one of the numerous types of quadrant electrometer. With the help of auxiliary capacities, the electrometer can be used to measure currents with accuracy over a wide range, and can be employed for practically every kind of measurement required in radio-activity.
The elementary theory of the symmetrical quadrant electrometer as given in the text-books is very imperfect. It is deduced that the sensibility of the electrometer—measured by the deflection of the needle for 1 volt P.D. between the quadrants—varies directly as the potential of the charged needle, provided that this potential is high compared with the P.D. between the quadrants. In most electrometers however, the sensibility rises to a maximum, and then decreases with increase of potential of the needle. For electrometers in which the needle lies close to the quadrants, this maximum sensibility is obtained for a comparatively low potential of the needle. A theory of the quadrant electrometer, accounting for this action, has been recently given by G. W. Walker[104]. The effect appears to be due to the presence of the air space that necessarily exists between adjoining quadrants.
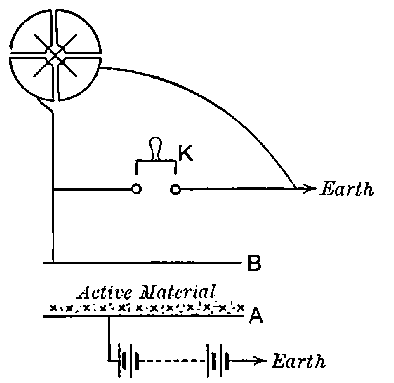
Fig. 14.
Suppose that it is required to measure with an electrometer the ionization current between two horizontal metal plates A and B (Fig. 14) on the lower of which some active material has been spread. If the saturation current is required, the insulated plate A is connected with one pole of a battery of sufficient E.M.F. to produce saturation, the other pole being connected to earth. The insulated plate B is connected with one pair of quadrants of the electrometer, the other pair being earthed. By means of a suitable key K, the plate B and the pair of quadrants connected with it may be 91either insulated or connected with earth. When a measurement is to be taken, the earth connection is broken. If the positive pole of the battery is connected with A, the plate B and the electrometer connections immediately begin to be charged positively, and the potential, if allowed, will steadily rise until it is very nearly equal to the potential of A. As soon as the potential of the electrometer system begins to rise, the electrometer needle commences to move at a uniform rate. Observations of the angular movement of the needle are made either by the telescope and scale or by the movement of the spot of light on a scale in the usual way. If the needle is damped so as to give a uniform motion over the scale, the rate of movement of the needle, i.e. the number of divisions of the scale passed over per second, may be taken as a measure of the current through the gas. The rate of movement is most simply obtained by observing with a stop-watch the time taken for the spot of light, after the motion has become steady, to pass over 100 divisions of the scale. As soon as the observation is made, the plate B is again connected with earth, and the electrometer needle returns to its original position.
In most experiments on radio-activity, only comparative measurements of saturation currents are required. If these measurements are to extend over weeks or months, as is sometimes the case, it is necessary to adopt some method of standardizing the electrometer from day to day, so as to correct for variation in its sensibility. This is done most simply by comparing the current to be measured with that due to a standard sample of uranium oxide, which is placed in a definite position in a small testing vessel, always kept in connection with the electrometer. Uranium oxide is a very constant source of radiation, and the saturation current due to it is the same from day to day. By this method of comparison accurate observations may be made on the variation of activity of a substance over long intervals of time, although the sensibility of the electrometer may vary widely between successive measurements.
60. Construction of electrometers. As the quadrant electrometer has gained the reputation of being a difficult and uncertain instrument for accurate measurements of current, it may 92be of value to give some particular details in regard to the best method of construction and insulation. In most of the older types of quadrant electrometers the needle system was made unnecessarily heavy. In consequence of this, if a sensibility of the order of 100 mms. deflection for 1 volt was required, it was necessary to charge the Leyden jar connected to the needle to a fairly high potential. This at once introduced difficulties, for at a high potential it is not easy to insulate the Leyden jar satisfactorily, or to charge it to the same potential from day to day. This drawback is to a large extent avoided in the White pattern of the Kelvin electrometer, which is provided with a replenisher and attracted disc for keeping the potential of the needle at a definite value. If sufficient trouble is taken in insulating and setting up this type of electrometer, it proves a very useful instrument of moderate sensibility, and will continue in good working order for a year or more without much attention.
Simpler types of electrometer of greater sensibility can however be readily constructed to give accurate results. The old type of quadrant electrometer, to be found in every laboratory, can readily be modified to prove a useful and trustworthy instrument. A light needle can be made of thin aluminium, of silvered paper or of a thin plate of mica, covered with gold-leaf to make it conducting. The aluminium wire and mirror attached should be made as light as possible. The needle should be supported either by a fine quartz fibre or a long bifilar suspension of silk. A very fine phosphor bronze wire of some length is also very satisfactory. A magnetic control is not very suitable, as it is disturbed by coils or dynamos working in the neighbourhood. In addition, the zero point of the needle is not as steady as with the quartz or bifilar suspension.
When an electrometer is used to measure a current by noting the rate of movement of the needle, it is essential that the needle should be damped sufficiently to give a uniform motion of the spot of light over the scale. The damping requires fairly accurate adjustment. If it is too little, the needle has an oscillatory movement superimposed on the steady motion; if it is too great, it moves too sluggishly from rest and takes some time to attain a state of uniform motion. With a light needle, very little, if 93any, extra damping is required. A light platinum wire with a single loop dipping in sulphuric acid is generally sufficient for the purpose.
With light needle systems and delicate suspensions, it is only necessary to charge the needle to a potential of a few hundred volts to give a sensibility of several thousand divisions for a volt. With such low potentials, the difficulty of insulation of the condenser, with which the needle is in electrical connection, is much reduced. It is convenient to use a condenser such that the potential of the needle does not fall more than a few per cent. per day. The ordinary short glass jar partly filled with sulphuric acid is, in most cases, not easy to insulate to this extent. It is better to replace it by an ebonite (or sulphur) condenser[105] such as is shown in Fig. 15.
Fig. 15.
A circular plate of ebonite about 1 cm. thick is turned down until it is not more than ½ mm. thick in the centre. Into this circular recess a brass plate B fits loosely. The ebonite plate rests on another brass plate C connected with earth. The condenser thus formed has a considerable capacity and retains a charge for a long time. In order to make connection with the needle, a small glass vessel D, partly filled with sulphuric acid, is placed on the plate B and put in connection with the needle by means of a fine platinum wire. The platinum wire from the needle dips into the acid, and serves to damp the needle. In a dry atmosphere, a condenser of this kind will not lose more than 20 per cent. of its charge in a week. If the insulation deteriorates, it can readily be made good by rubbing the edge of the ebonite A with sand-paper, or removing its surface in a lathe.
If a sufficient and steady E.M.F. is available, it is much better to keep the battery constantly connected with the needle, and to 94avoid the use of the condenser altogether. If a battery of small accumulators is used, their potential can be kept at a constant value, and the electrometer always has a constant sensibility.
61. A very useful electrometer of great sensibility has been devised by Dolezalek[106]. It is of the ordinary quadrant type with a very light needle of silvered paper, spindle shaped, which lies fairly close to the quadrants. A very fine quartz suspension is employed. In consequence of the lightness of the needle and its nearness to the quadrants, it acts as its own damper. This is a great advantage, for difficulties always arise when the wire dips into sulphuric acid, on account of the thin film which collects after some time on the surface of the acid. This film obstructs the motion of the platinum wire dipping into the acid, and has to be removed at regular intervals. These instruments can readily be made to give a sensibility of several thousand divisions for a volt when the needle is charged to about one hundred volts. The sensibility of the electrometer passes through a maximum as the potential of the needle is increased. It is always advisable to charge the needle to about the value of this critical potential. The capacity of the electrometer is in general high (about 50 electrostatic units) but the increased sensibility more than compensates for this. The needle may either be charged by lightly touching it with one terminal of a battery, or it may be kept charged to a constant potential through the quartz suspension.
Dolezalek states that the fibre can be made sufficiently conducting for the purpose by dipping it into a dilute solution of calcium chloride or phosphoric acid. I have not found this method satisfactory in dry climates as in many cases the fibre practically loses its conductivity after a few days exposure to dry air.
In addition to its great sensibility, the advantage of this instrument is in the steadiness of the zero and in the self-damping.
A sensibility of 10,000 millimetre divisions per volt can be readily obtained with this electrometer, if a very fine fibre be used. The use of such high sensibilities cannot, however, be recommended except for very special experiments. The period of swing of the needle under these conditions is several minutes and the natural 95leak of the testing vessels employed, as well as electrostatic and other disturbances, make themselves only too manifest. If measurements of minute currents are required, an electroscope of the type described in Section 56 is much to be preferred to a very sensitive electrometer. The electroscope readings in such a case are more accurate than similar measurements made by an electrometer.
For most measurements in radio-activity, an electrometer which has a sensibility of 100 divisions per volt is very suitable, and no advantage is gained by using an electrometer of greater sensibility. If still smaller effects require to be measured, the sensibility may be increased to several thousand divisions per volt.
62. Adjustment and screening. In adjusting an electrometer, it is important to arrange that the needle shall lie symmetrically with regard to the quadrants. This is best tested by observing whether the needle is deflected on charging, the quadrants all being earthed. In most electrometers there is an adjustable quadrant, the position of which may be altered until the needle is not displaced on charging. When this condition is fulfilled, the zero reading of the electrometer remains unaltered as the needle loses its charge, and the deflection on both sides of the zero should be the same for equal and opposite quantities of electricity.
The supports of the quadrants require to be well insulated. Ebonite rods are as a rule more satisfactory for this purpose than glass. In testing for the insulation of the quadrants and the connections attached, the system is charged to give a deflection of about 200 scale divisions. If the needle does not move more than one or two divisions after standing for one minute, the insulation may be considered quite satisfactory. When a suitable desiccator is placed inside the tight-fitting electrometer case, the insulation of the quadrants should remain good for months. If the insulation of the ebonite deteriorates, it can easily be made good by removing the surface of the ebonite in a lathe.
In working with a sensitive instrument like the Dolezalek electrometer, it is essential that the electrometer and the testing apparatus should be completely enclosed in a screen of wire-gauze connected with earth, in order to avoid electrostatic disturbances. 96If an apparatus is to be tested at some distance from the electrometer, the wires leading to it should be insulated in metal cylinders connected with earth. The size of the insulators used at various points should be made as small as possible, in order to avoid disturbances due to their electrification. In damp climates, paraffin, amber, or sulphur insulates better than ebonite. The objection to paraffin as an insulator for sensitive electrometers lies in the difficulty of getting entirely rid of any electrification on its surface. When paraffin has been once charged, the residual charge, after diselectrifying it with a flame, continues to leak out for a long interval. All insulators should be diselectrified by means of a spirit-lamp or still better by leaving some uranium near them. Care should be taken not to touch the insulation when once diselectrified.
In accurate work it is advisable to avoid the use of gas jets or Bunsen flames in the neighbourhood of the electrometer, as the flame gases are strongly ionized and take some time to lose their conductivity. If radio-active substances are present in the room, it is necessary to enclose the wires leading to the electrometer in fairly narrow tubes, connected with earth. If this is not done, it will be found that the needle does not move at a constant rate, but rapidly approaches a steady deflection where the rate of loss of charge of the electrometer and connections, due to the ionization of the air around them, is balanced by the current to be measured. This precaution must always be taken when observations are made on the very penetrating rays from active substances. These rays readily pass through ordinary screens, and ionize the air around the electrometer and connecting wires. For this reason it is impossible to make accurate measurements of small currents in a room which is used for the preparation of radio-active material. In course of time the walls of the room become radio-active owing to the dissemination of dust and the action of the radio-active emanations[107].
9763. Electrometer key. For work with electrometers of high sensibility, a special key is necessary to make and break from a distance the connection of the quadrants with earth in order to avoid electrostatic disturbances at the moment the current is to be measured. The simple key shown in Fig. 16 has been found very satisfactory for this purpose. A small brass rod BM, to which a string is attached, can be moved vertically up and down in a brass tube A, which is rigidly attached to a bent metal support connected with earth. When the string is released, this rod makes contact with the mercury M, which is placed in a small metal vessel resting on a block of ebonite P. The electrometer and testing vessel are connected with the mercury. When the string is pulled, the rod BM is removed from the mercury and the earth connection of the electrometer system is broken. On release of the string, the rod BM falls and the electrometer is again earthed. By means of this key, which may be operated at any distance from the electrometer, the earth connection may be made and broken at definite intervals without any appreciable disturbance of the needle.
Fig. 16.
64. Testing apparatus. The arrangement shown in Fig. 17 is very convenient for many measurements in radio-activity. Two parallel insulated metal plates A and B are placed inside a metal vessel V, provided with a side door. The plate A is connected with one terminal of a battery of small storage cells, the other pole of which is earthed; the plate B with the electrometer, and the vessel V with earth. The shaded areas in the figure indicate the position of ebonite insulators. The active material to be tested is spread uniformly in a shallow groove (about 5 cms. square and 2 mms. deep) in the brass plate A. In order to avoid breaking the battery connection every time the plate A is removed, the wire from the battery is permanently connected with the metal block N 98resting on the ebonite support. In this arrangement there is no possibility of a conduction leak from the plate A to B, since the earth-connected vessel V intervenes.
Fig. 17.
An apparatus of this kind is very convenient for testing the absorption of the radiations by solid screens, as well as for making comparative studies of the activity of different bodies. Unless very active preparations of radium are employed, a battery of 300 volts is sufficient to ensure saturation when the plates are not more than 5 centimetres apart. If substances which give off a radio-active emanation are being tested, the effect of the emanation can be eliminated by passing a steady current of air from a gas bag between the plates. This removes the emanation as fast as it is produced.
If a clean plate is put in the place of A, a small movement of the electrometer needle is always observed. If there is no radio-active substance in the neighbourhood, this effect is due to the small natural ionization of the air. We can correct for this natural leak when necessary.
65. We have often to measure the activity due to the emanations of thorium or radium, or the excited activity produced by those emanations on rods or wires. A convenient apparatus for this purpose is shown in Fig. 18. The cylinder B is connected with 99the battery in the usual way, and the central conductor A with the electrometer. This central rod is insulated from the external cylinder by an ebonite cork, which is divided into two parts by a metal ring CC´ connected to earth. This ring acts the part of a guard-ring, and prevents any conduction leak between B and A. The ebonite is thus only required to insulate satisfactorily for the small rise of potential produced on A during the experiment. In all accurate measurements of current in radio-activity the guard-ring principle should always be used to ensure good insulation. This is easily secured when the ebonite is only required to insulate for a fraction of a volt, instead of for several hundred volts, as is the case when the guard-ring is absent.
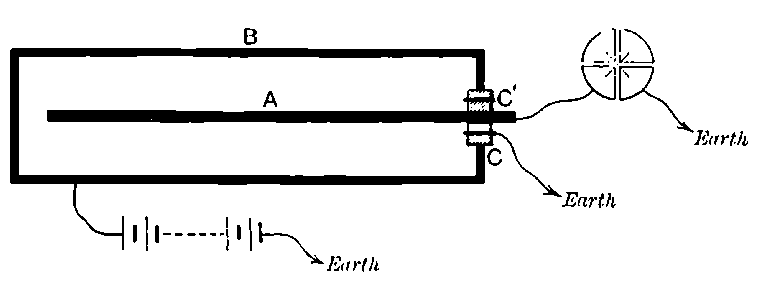
Fig. 18.
66. For measurements of radio-activity with an electrometer, a steady source of E.M.F. of at least 300 volts is necessary. This is best obtained by a battery of small cells simply made by immersing strips of lead in dilute sulphuric acid, or by a battery of small accumulators of the usual construction. Small accumulators of capacity about one-half ampere-hour can now be obtained at a moderate price, and are more constant and require less attention than simple lead cells.
In order to measure currents over a wide range, a graduated series of capacities is required. The capacity of an electrometer and testing apparatus is usually about 50 electrostatic units or ·000056 microfarads. Subdivided condensers of mica are constructed in which capacities varying from ·001 to ·2 microfarads are provided. With such a condenser, another extra capacity is required to bridge over the gap between the capacity of the electrometer and 100the lowest capacity of the condenser. This capacity of value about 200 electrostatic units can readily be made by using parallel plates or still better concentric cylinders. With this series of capacities, currents may be measured between 3 × 10-14 and 3 × 10-8 amperes—a range of over one million. Still larger currents can be measured if the sensibility of the electrometer is reduced, or if larger capacities are available.
In a room devoted to electrometer measurements of radio-activity, it is desirable to have no radio-active matter present except that to be tested. The room should also be as free from dust as possible. The presence of a large quantity of dust in the air (see section 31) is a very disturbing factor in all radio-active measurements. A larger E.M.F. is required to produce saturation on account of the diffusion of the ions to the dust particles. The presence of dust in the air also leads to uncertainty in the distribution of excited activity in an electric field (see section 181).
67. Measurement of Current. In order to determine the current in the electrometer circuit by measuring the rate of movement of the needle, it is necessary to know both the capacity of the circuit and the sensibility of the electrometer.
The current i is given by the product of the capacity of the system and the rate of rise of potential.
Suppose, for example,
Since the electrometer can readily measure a current corresponding to a movement of half a scale division per second, we see that an electrometer can measure a current of 3 × 10-14 101amperes, which is considerably below the range of the most sensitive galvanometer.
The capacity of the electrometer itself must not be considered as equal to that of the pair of quadrants and the needle when in a position of rest. The actual capacity is very much larger than this, on account of the motion of the charged needle. Suppose, for example, that the needle is charged to a high negative potential, and kept at the zero position by an external constraint. If a quantity Q of positive electricity is given to the electrometer and its connections, the whole system is raised to a potential V, such that Q = CV, where C is the capacity of the system. When however the needle is allowed to move, it is attracted into the charged pair of quadrants. This corresponds to the introduction of a negatively charged body between the quadrants, and in consequence the potential of the system is lowered to V´. The actual capacity C´ of the system when the needle moves is thus greater than C, and is given by
Thus the capacity of the electrometer is not a constant, but depends on the potential of the needle, i.e. on the sensibility of the electrometer.
An interesting result of practical importance follows from the variation of the capacity of the electrometer with the potential of the needle. If the external capacity attached to the electrometer is small compared with that of the electrometer itself, the rate of movement of the needle for a constant current is, in some cases, independent of the sensibility. An electrometer may be used for several days or even weeks to give nearly equal deflections for a constant current, without recharging the needle, although its potential has been steadily falling during the interval. In such a case the decrease in sensibility is nearly proportional to the decrease in capacity of the electrometer, so that the deflection for a given current is only slightly altered. The theory of this action has been given by J. J. Thomson[108].
68. The capacity of the electrometer and its connections cannot be measured by any of the commutator methods used for the determination of small capacities, for in such cases the needle 102does not move, and the capacity measured is not that of the electrometer system when in actual use. The value of the capacity may, however, be determined by the method of mixtures.
The electrometer and its connections are charged to a potential V1 by a battery, and the deflection d1 of the needle is noted. By means of an insulated key, the capacity of the standard condenser is added in parallel with the electrometer system. Let V2 be the potential of the system, and d2 the new deflection.
Then
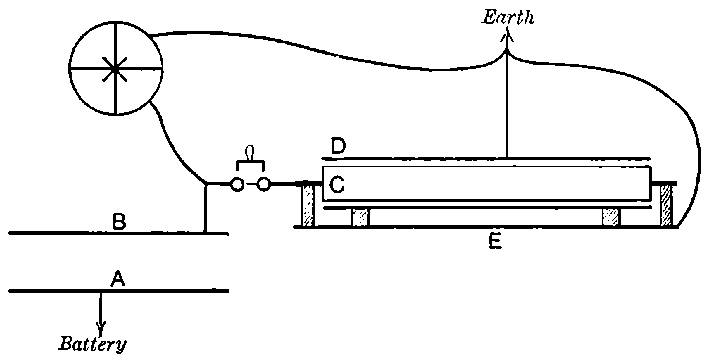
Fig. 19.
A simple standard capacity for this purpose can be constructed of two concentric brass tubes the diameters of which can be accurately measured. The external cylinder D (Fig. 19) is mounted on a wooden base, which is covered with a sheet of metal or tinfoil connected to earth. The tube C is supported centrally on ebonite rods at each end. The capacity is given approximately by the formula
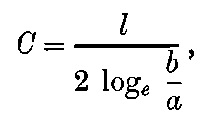
103where b is the internal diameter of D, a the external diameter of C, and l the length of the tubes.
The following method can be used in some cases with advantage. While a testing vessel is in connection with the electrometer, a sample of uranium is placed on the lower plate A. Let d2 and d1 be the number of divisions passed over per second by the needle with and without the standard capacity in connection.
This method has the advantage that the relative capacities are expressed in terms of the motion of the needle under the actual conditions of measurement.
69. Steady deflection method. The methods of measurement previously described depend upon the rate of angular movement of a suspended gold-leaf or of an electrometer needle. The galvanometer can only be employed for measurements with intensely active matter. A need, however, has long been felt for a method in which ordinary ionization currents can be measured by means of a steady deflection of an electrometer needle. This is especially the case, where measurements have to be made with active substances whose activity alters rapidly in the course of a few minutes.
This can obviously be secured if the electrometer system (one pair of quadrants being earthed) is connected to earth through a suitable high resistance. A steady deflection of the electrometer needle will be obtained when the rate of supply of electricity to the electrometer system is balanced by the loss due to conduction through the resistance. If the high resistance obeys Ohm’s law, the deflection should be proportional to the ionization current to be measured.
A simple calculation shows that the resistance required is very great. Suppose, for example, that a current is to be measured corresponding to a rate of movement of the needle of 5 divisions per second, with a sensibility of 1000 divisions per volt, and where 104the capacity of the electrometer system is 50 electrostatic units. This current is equal to 2·8 × 10-13 amperes. If a steady deflection of 10 divisions is required, which corresponds to a rise of potential of the system of ¹⁄₁₀₀ of a volt, the resistance should be 36,000 megohms. For a deflection of 100 divisions, the resistance should be 10 times as large. Dr Bronson[109], working in the laboratory of the writer, has recently made some experiments in order to devise a practical method for measurements of this character. It is difficult to obtain sufficiently high and constant resistances to answer the purpose. Tubes of xylol had too great a resistance, while special carbon resistances were not sufficiently constant. The difficulty was finally got over by the use of what may be called an “air resistance.” The arrangement of the experiment is shown in Fig. 20.
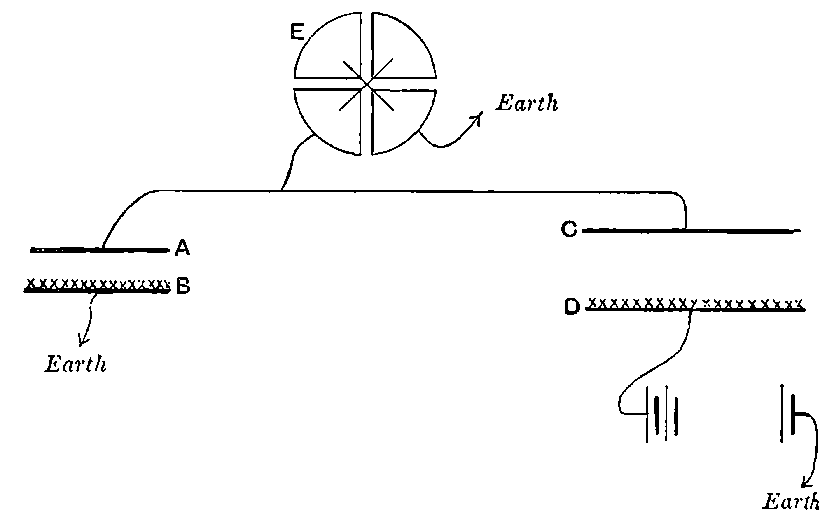
Fig. 20.
The electrometer system was connected with the upper of two insulated parallel plates AB, on the lower of which was spread a layer of a very active substance. An active bismuth plate, coated with radio-tellurium, which had been obtained from Sthamer of Hamburg, proved very convenient for this purpose.
The lower plate B was connected to earth. The charge communicated to the upper plate of the testing vessel CD and the electrometer system leaked away in consequence of the strong 105ionization between the plates AB, and a steady deflection was obtained when the rate of supply was equal to the rate of discharge.
This air resistance obeyed Ohm’s law over a considerable range, i.e. the steady deflection was proportional to the current. It is advisable, in such an arrangement, to test whether the deflection is proportional to the ionization current over the range required for measurement. This can readily be done by the use of a number of metal vessels filled with a constant radio-active substance like uranium oxide. The effect of these, when placed in the testing vessel, can be tested separately and in groups, and in this way the scale can be calibrated accurately.
The plates AB were placed inside a closed vessel to avoid air currents. The contact difference of potential between the plates AB, which shows itself by a steady deflection when no radio-active matter is present in CD, was for the most part eliminated by covering the surface of the plates A and B with very thin aluminium foil.
This method proved very accurate and convenient for measurement of rapid changes in activity, and possesses many advantages over the ordinary rate-method of use of an electrometer. A thin layer of radium of moderate activity would probably serve in place of the radio-tellurium, but the emanation and the β and γ rays emitted from it would be a possible source of disturbance to the measurements. The deflection of the electrometer needle in this arrangement is independent of the capacity of the electrometer system, and thus comparative measurements of current can be made without the necessity of determining the capacity in each case.
70. Quartz piezo-electrique. In measurements of the strength of currents by electrometers, it is always necessary to determine the sensibility of the instrument and the capacity of the electrometer and the apparatus attached thereto. By means of the quartz piezo-electrique devised by the brothers MM. J. and P. Curie[110], measurements of the current can be made with rapidity and accuracy over a wide range. These measurements are quite independent of the capacity of the electrometer and external circuit.
106The essential part of this instrument consists of a plate of quartz which is cut in a special manner. When this plate is placed under tension, there is a liberation of electricity equal in amount but opposite in sign on the two sides of the plate. The plate of quartz AB (Fig. 21) is hung vertically and weights are added to the lower end. The plate is cut so that the optic axis of the crystal is horizontal and at right angles to the plane of the paper.
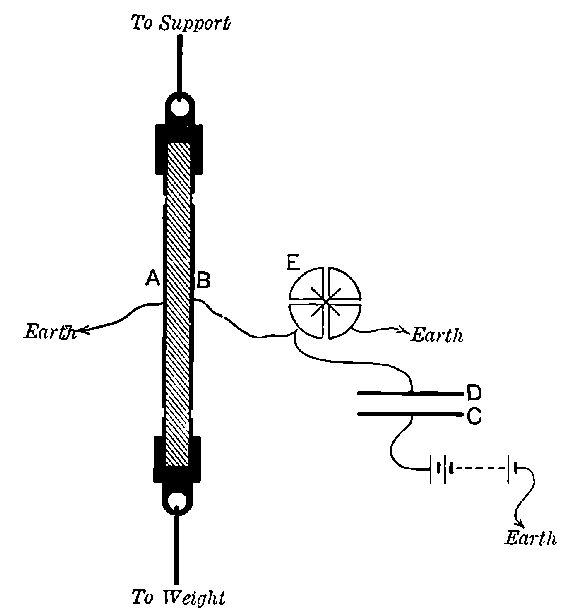
Fig. 21.
The two faces A and B are normal to one of the binary axes (or electrical axes) of the crystal. The tension must be applied in a direction normal to the optic and electric axes. The two faces A and B are silvered, but the main portion of the plate is electrically insulated by removing a narrow strip of the silvering near the upper and lower ends of the plate. One side of the plate is connected with the electrometer and with the conductor, the rate of leak of which is to be measured. The quantity of electricity set free on one face of the plate is accurately given by
107where L is the length of the insulated portion of the plate, b the thickness AB, and F the weight attached in kilogrammes. Q is then given in electrostatic units.
Suppose, for example, that it is required to measure the current between the plates CD (Fig. 21) due to some radio-active material on the plate C, for a given difference of potential between C and D. At a given instant the connection of the quadrants of the electrometer with the earth is broken. The weight is attached to the quartz plate, and is held in the hand so as to apply the tension gradually. This causes a release of electricity opposite in sign to that given to the plate D. The electrometer needle is kept at the position of rest as nearly as possible by adjusting the tension by hand. The tension being fully applied, the moment the needle commences to move steadily from zero is noted. The current between the plates CD is then given by Q/t where t is the time of the observation. The value of Q is known from the weight attached.
In this method the electrometer is only used as a detector to show that the system is kept at zero potential. No knowledge of the capacity of the insulated system is required. With practice, measurements of the current can be made in this way with rapidity and certainty.
71. The Three Types of Radiation. All the radio-active substances possess in common the power of acting on a photographic plate and of ionizing the gas in their immediate neighbourhood. The intensity of the radiations may be compared by means of their photographic or electrical action; and, in the case of the strongly radio-active substances, by the power they possess of lighting up a phosphorescent screen. Such comparisons, however, do not throw any light on the question whether the radiations are of the same or of different kinds, for it is well known that such different types of radiations as the short waves of ultra-violet light, Röntgen and cathode rays, all possess the property of producing ions throughout the volume of a gas, lighting up a fluorescent screen, and acting on a photographic plate. Neither can the ordinary optical methods be employed to examine the radiations under consideration, as they show no trace of regular reflection, refraction, or polarization.
Two general methods can be used to distinguish the types of the radiations given out by the same body, and also to compare the radiations from the different active substances. These methods are as follows:
(1) By observing whether the rays are appreciably deflected in a magnetic field.
(2) By comparing the relative absorption of the rays by solids and gases.
Examined in these ways, it has been found that there are three different types of radiation emitted from radio-active bodies, which 109for brevity and convenience have been termed by the writer the α, β, and γ rays.
(i) The α rays are very readily absorbed by thin metal foil and by a few centimetres of air. They have been shown to consist of positively charged bodies projected with a velocity of about ⅒ the velocity of light. They are deflected by intense magnetic and electric fields, but the amount of deviation is minute in comparison with the deviation, under the same conditions, of the cathode rays produced in a vacuum tube.
(ii) The β rays are far more penetrating in character than the α rays, and consist of negatively charged bodies projected with velocities of the same order as the velocity of light. They are far more readily deflected than the α rays, and are in fact identical with the cathode rays produced in a vacuum tube.
(iii) The γ rays are extremely penetrating, and non-deviable by a magnetic field. Their true nature is not definitely settled, but they are analogous in most respects to very penetrating Röntgen rays.
The three best known radio-active substances, uranium, thorium, and radium, all give out these three types of rays, each in an amount approximately proportional to its relative activity measured by the α rays. Polonium stands alone in giving only the α or easily absorbed rays[111].
72. Deflection of the rays. The rays emitted from the active bodies thus present a very close analogy with the rays which are produced in a highly exhausted vacuum tube when an electric 110discharge passes through it. The α rays correspond to the canal rays, discovered by Goldstein, which have been shown by Wien to consist of positively charged bodies projected with great velocity (see section 51). The β rays are the same as the cathode rays, while the γ rays resemble the Röntgen rays. In a vacuum tube, a large amount of electric energy is expended in producing the rays, but, in the radio-active bodies, the rays are emitted spontaneously, and at a rate uninfluenced by any chemical or physical agency. The α and β rays from the active bodies are projected with much greater velocity than the corresponding rays in a vacuum tube, while the γ rays are of much greater penetrating power than Röntgen rays.
The effect of a magnetic field on a pencil of rays from a radio-active substance giving out the three kinds of rays is very well illustrated in Fig. 22[112].
Fig. 22.
Some radium is placed in the bottom of a narrow cylindrical lead vessel R. A narrow pencil of rays consisting of α, β, and γ rays escapes from the opening. If a strong uniform magnetic field is applied at right angles to the plane of the paper, and directed towards the paper, the three types of rays are separated from one another. The γ rays continue in a straight line without any deviation. The β rays are deflected to the right, describing circular orbits the radii of which vary within wide limits. If the photographic plate AC is placed under the radium vessel, the β rays produce a diffuse photographic impression on the right of the vessel R. The α rays are bent in the direction opposite to that of the β rays, and describe a portion of the arc of a circle of large radius, but they are rapidly absorbed after traversing a distance of a few centimetres from the vessel R. The amount 111of the deviation of the α rays compared with that of the β rays is much exaggerated in the figure.
73. Ionizing and penetrating power of the rays. Of the three kinds of rays, the α rays produce most of the ionization in the gas and the γ rays the least. With a thin layer of unscreened active material spread on the lower of two parallel plates 5 cms. apart, the amount of ionization due to the α, β, and γ rays is of the relative order 10,000, 100, and 1. These numbers are only rough approximations, and the differences become less marked as the thickness of the radio-active layer increases.
The average penetrating power of the rays is shown below. In the first column is given the thickness of the aluminium, which cuts each radiation down to half its value, and in the second the relative power of penetration of the rays.
| Radiation | Thickness of Aluminium in cms. which cuts off half the radiation | Relative power of penetration |
|---|---|---|
| α rays | 0·0005 cms. | 1 |
| β „ | 0·05 cms. | 100 |
| γ „ | 8 cms. | 10000 |
The relative power of penetration is thus approximately inversely proportional to the relative ionization. These numbers, however, only indicate the order of relative penetrating power. This power varies considerably for the different active bodies.
The α rays from uranium and polonium are the least penetrating, and those from thorium the most. The β radiations from thorium and radium are very complex, and consist of rays widely different in penetrating power. Some of the β rays from these substances are much less and others much more penetrating than those from uranium, which gives out fairly homogeneous rays.
74. Difficulties of comparative measurements. It is difficult to make quantitative or even qualitative measurements of the relative intensity of the three types of rays from active substances. The three general methods employed depend upon the action of the rays in ionizing the gas, in acting on a photographic 112plate, and in causing phosphorescent or fluorescent effects in certain substances. In each of these methods the fraction of the rays which is absorbed and transformed into another form of energy is different for each type of ray. Even when one specific kind of ray is under observation, comparative measurements are rendered difficult by the complexity of that type of rays. For example, the β rays from radium consist of negatively charged particles projected with a wide range of velocity, and, in consequence, they are absorbed in different amounts in passing through a definite thickness of matter. In each case, only a fraction of the energy absorbed is transformed into the particular type of energy, whether ionic, chemical, or luminous, which serves as a means of measurement.
The rays which are the most active electrically are the least active photographically. Under ordinary conditions, most of the photographic action of uranium, thorium, and radium, is due to the β or cathodic rays. The α rays from uranium and thorium, on account of their weak action, have not yet been detected photographically. With active substances like radium and polonium, the α rays readily produce a photographic impression. So far the γ rays have been detected photographically from radium only. That no photographic action of these rays has yet been established for uranium and thorium is probably due merely to the fact that the effect sought for is very small, and during exposures for long intervals it is very difficult to avoid fogging of the plates owing to other causes. Considering the similarity of the radiations in other respects, there can be little doubt that the γ rays do produce some photographic action, though it is too small to observe with certainty.
These differences in the photographic and ionizing properties of the radiations must always be taken into account in comparing results obtained by the two methods. The apparent contradiction of results obtained by different observers using these two methods is found to be due to their differences in relative photographic and ionizing action. For example, with the unscreened active material, the ionization observed by the electrical method is due almost entirely to α rays, while the photographic action under the same condition is due almost entirely to the β rays.
It is often convenient to know what thickness of matter is sufficient to absorb a specific type of radiation. A thickness of 113aluminium or mica of ·01 cms. or a sheet of ordinary writing-paper is sufficient to absorb completely all the α rays. With such a screen over the active material, the effects are due only to the β and γ rays, which pass through with a very slight absorption. Most of the β rays are absorbed in 5 mms. of aluminium or 2 mms. of lead. The radiation passing through such screens consists very largely of the γ rays. As a rough working rule, it may be taken that a thickness of matter required to absorb any type of rays is inversely proportional to the density of the substance, i.e. the absorption is proportional to the density. This rule holds approximately for light substances, but, in heavy substances like mercury and lead, the radiations are about twice as readily absorbed as the density rule would lead us to expect.
75. Discovery of the β rays. A discovery which gave a great impetus to the study of the radiations from active bodies was made in 1899, almost simultaneously in Germany, France, and Austria. It was observed that preparations of radium gave out some rays which were deviable by a magnetic field, and very similar in character to the cathode rays produced in a vacuum tube. The observation of Elster and Geitel that a magnetic field altered the conductivity produced in air by radium rays, led Giesel[113] to examine the effect of a magnetic field on the radiations. In his experiments, the radio-active preparation was placed in a small vessel between the poles of an electromagnet. The vessel was arranged to give a pencil of rays which was approximately perpendicular to the field. The rays caused a small fluorescent patch on the screen. On exciting the electromagnet, the fluorescent zone was observed to broaden out on one side. On reversing the field, the extension of the zone was in the opposite direction. The deviation of the rays thus indicated was in the same direction and of the same order of magnitude as that for cathode rays.
S. Meyer and Schweidler[114] also obtained similar results. They 114showed, in addition, the deviation of the rays by the alteration of the conductivity of the air when a magnetic field was applied. Becquerel[115], a little later, showed the magnetic deflection of the radium rays by using the photographic method. P. Curie[116], by the electrical method, showed furthermore that the rays from radium consisted of two kinds, one apparently non-deviable and easily absorbed (now known as the α rays), and the other penetrating and deviable by a magnetic field (now known as the β rays). The ionization effect due to the β rays was only a small fraction of that due to the α rays. At a later date Becquerel, by the photographic method, showed that uranium gave out some deflectable rays. It had been shown previously[117] that the rays from uranium consisted of α and β rays. The deflected rays in Becquerel’s experiment consisted entirely of β rays, as the α rays from uranium produce no appreciable photographic action. Rutherford and Grier[118], using the electric method, showed that compounds of thorium, like those of uranium, gave out, besides α rays, some penetrating β rays, deviable in a magnetic field. As in the case of radium, the ionization due to the α rays of uranium and thorium is large compared with that due to the β rays.
76. Examination of the magnetic deviation by the photographic method. Becquerel has made a very complete study, by the photographic method, of the β rays from radium, and has shown that they behave in all respects like cathode rays, which are known to be negatively charged particles moving with a high velocity. The motion of a charged ion acted on by a magnetic field has been discussed in section 49. It has been shown that if a particle of mass m and charge e is projected with a velocity u, at an angle α with the direction of a uniform field of strength H, it will describe a helix round the magnetic lines of force. This helix is wound on a cylinder of radius R, with the axis parallel to the field, where R is given by
115When α = π/2, i.e. when the rays are projected normally to the field, the particles describe circles of radius
The planes of these circles are normal to the field. Thus, for a particular velocity u, the value of R varies inversely as the strength of the field. In a uniform field the rays projected normally to the field describe circles, and their directions of projection are the tangents at the origin.
This conclusion has been verified experimentally by Becquerel for the β rays of radium, by an arrangement similar to that shown in Fig. 23.
Fig. 23.
A photographic plate P, with the film downwards, is enveloped in black paper and placed horizontally in the uniform horizontal magnetic field of an electromagnet. The magnetic field is supposed to be uniform, and, in the figure, is at right angles to the plane of the paper. The plate was covered with a sheet of lead, and on the edge of the plate, in the centre of the magnetic field, is placed a small lead vessel R containing the radio-active matter.
On exciting the magnet, so that the rays are bent to the left of the figure, it is observed that a photographic impression is produced directly below the source of the rays, which have been bent round by the magnetic field. The active matter sends out rays equally in all directions. The rays perpendicular to the field describe circles, which strike the plate immediately under the source. A few of these rays, A1, A2, A3, are shown in the figure. The rays, normal to the plate, strike the plate almost normally, 116while the rays nearly parallel to the plate strike the plate at grazing incidence. The rays, inclined to the direction of the field, describe spirals and produce effects on an axis parallel to the field passing through the source. In consequence of this, any opaque screen placed in the path of the rays has its shadow thrown near the edge of the photographic plate.
77. Complexity of the rays. The deviable rays from radium are complex, i.e. they are composed of a flight of particles projected with a wide range of velocity. In a magnetic field every ray describes a path, of which the radius of curvature is directly proportional to the velocity of projection. The complexity of the radiation has been shown very clearly by Becquerel[119] in the following way.
An uncovered photographic plate, with the film upwards, was placed horizontally in the horizontal uniform magnetic field of an electromagnet. A small, open, lead box, containing the radio-active matter, was placed in the centre of the field, on the photographic plate. The light, due to the phosphorescence of the radio-active matter, therefore, could not reach the plate. The whole apparatus was placed in a dark room. The impression on the plate took the form of a large, diffuse, but continuous band, elliptic in shape, produced on one side of the plate.
Such an impression is to be expected if the rays are sent out in all directions, even if their velocities of projection are the same, for it can readily be shown theoretically, that the path of the rays is confined within an ellipse whose minor axis, which is at right angles to the field, is equal to 2R, and whose major axis is equal to πR. If, however, the active matter is placed in the bottom of a deep lead cylinder of small diameter, the rays have practically all the same direction of projection, and in that case each part of the plate is acted on by rays of a definite curvature.
In this case also, a diffuse impression is observed on the plate, giving, so to speak, a continuous spectrum of the rays and showing that the radiation is composed of rays of widely different curvatures. Fig. 24 shows a photograph of this kind obtained by Becquerel, with strips of paper, aluminium, and platinum placed on the plate.

Fig. 24.
If screens of various thickness are placed on the plate, it is observed that the plate is not appreciably affected within a certain distance from the active matter, and that this distance increases with the thickness of the screen. This distance is obviously equal to twice the radius of curvature of the path of the rays, which are just able to produce an impression through the screen.
These experiments show very clearly that the most deviable rays are those most readily absorbed by matter. By observations of this kind Becquerel has determined approximately the inferior limit of the value of HR for rays which are transmitted through different thicknesses of matter.
The results are given in the table below:
| Substance | Thickness in mms. | Inferior limit of HR for transmitted rays |
|---|---|---|
| Black paper | 0·065 | 650 |
| Aluminium | 0·010 | 350 |
| “ | 0·100 | 1000 |
| ” | 0·200 | 1480 |
| Mica | 0·025 | 520 |
| Glass | 0·155 | 1130 |
| Platinum | 0·030 | 1310 |
| Copper | 0·085 | 1740 |
| Lead | 0·130 | 2610 |
If e/m is a constant for all the rays, the value of HR is proportional to the velocity of the rays, and it follows from the table that the velocity of the rays which just produce an effect on the plate through ·13 mms. of lead is about 7 times that of the rays which 118just produce an impression through ·01 mm. of aluminium. It will be shown, however, in section 82, that e/m is not a constant for all speeds, but decreases with increase of velocity of the rays. The difference in velocity between the rays is in consequence not as great as this calculation would indicate. On examination of the rays from uranium, Becquerel found that the radiation is not as complex as that from radium, but consists wholly of rays for which the value of HR is about 2000.
78. Examination of the β rays by the electric method. The presence of easily deviable rays given off from an active substance can most readily be shown by the photographic method, but it is necessary, in addition, to show that the penetrating rays which produce the ionization in the gas are the same as those which cause the photographic action. This can be conveniently tested in an arrangement similar to that shown in Fig. 25.
Fig. 25.
The radio-active matter A is placed on a lead block B´´ between the two parallel lead plates BB´. The rays pass between the parallel plates and ionize the gas between the plates PP´ of the testing vessel. The magnetic field is applied at right angles to the plane of the paper. The dotted rectangle EEEE represents the position of the pole piece. If a compound of radium or thorium is under investigation, a stream of air is required to prevent the diffusion of the radio-active emanations into the testing vessel. When a layer of uranium, thorium or radium compound is placed at A, the ionization in the testing vessel is due mainly to the action of the α and β rays. The α rays are cut off by adding a layer of aluminium ·01 cm. thick over the active material. When the layer of active matter is not more than a few millimetres thick, the ionization due to the γ rays is small compared with that produced by the β rays, and may be neglected. On the application of a magnetic field at right angles to the mean 119direction of the rays, the ionization in the testing vessel due to the rays steadily decreases as the strength of the field increases, and in a strong field it is reduced to a very small fraction of its original value. In this case the rays are bent so that none of them enter the testing vessel.
Examined in this way, it has been found that the β rays of uranium, thorium, and radium consist entirely of rays readily deflected by a magnetic field. The rays from polonium consist entirely of α rays, the deviation of which can be detected only in very intense magnetic fields.
When the screen covering the active material is removed, in a strong magnetic field, the ionization in the vessel is mainly due to the α rays. On account of the slight deviation of the α rays under ordinary experimental conditions, a still greater increase of the magnetic field does not appreciably alter the current due to them in the testing vessel.
The action of a magnetic field on a very active substance like radium is easily shown by the electrical method, as the ionization current due to the deviable rays is large. With substances of small activity like uranium and thorium, the ionization current due to the deviable rays is very small, and a sensitive electrometer or an electroscope is required to determine the variation, in a magnetic field, of the very small current involved. This is especially the case for thorium oxide, which gives out only about ⅕ of the amount of deviable rays given out by the same weight of uranium oxide.
79. Experiments with a fluorescent screen. The β rays from a few milligrams of pure radium bromide produce intense fluorescence in barium platinocyanide and other substances which can be made luminous under the influence of the cathode rays. Using a centigram of radium bromide, the luminosity on a screen, placed upon it, is bright enough to be observed in daylight. With the aid of such a screen in a dark room many of the properties of the β rays may be simply illustrated and their complex nature clearly shown. A small quantity of radium is placed in the bottom of a short, narrow, lead tube open at one end. This is placed between the pole pieces of an electromagnet, and 120the screen placed below it. With no magnetic field, a faint luminosity of the screen is observed due to the very penetrating γ rays which readily pass through the lead. When the magnetic field is put on, the screen is brightly lighted up on one side over an area elliptical in shape (section 77). The direction of deviation is reversed by reversal of the field. The broad extent of the illumination shows the complex nature of the β rays. On placing a metallic object at various points above the screen, the trajectory of the rays can readily be traced by noticing the position of the shadow cast upon the screen. By observing the density of the shadow, it can be seen that the rays most easily deviated are the least penetrating.
80. Means of comparison. In order to prove the identity of the β rays from active bodies with the cathode rays produced in a vacuum tube, it is necessary to show
(1) That the rays carry with them a negative charge;
(2) That they are deviated by an electric as well as by a magnetic field;
(3) That the ratio e/m is the same as for the cathode rays.
Electric charge carried by the β rays. The experiments of Perrin and J. J. Thomson have shown that the cathode rays carry with them a negative charge. In addition, Lenard has shown that the rays still carry a charge after traversing thin layers of matter. When the rays are absorbed, they give up their charge to the body which absorbs them. The total amount of charge carried by the β rays from even a very active preparation of radium is, in general, small compared with that carried by the whole of the cathode rays in a vacuum tube, and can be detected only by delicate methods.
Suppose that a layer of very active radium is spread on a metal plate connected to earth, and that the β rays are absorbed by a parallel plate connected with an electrometer. If the rays are negatively charged, the top plate should receive a negative charge increasing with the time. On account, however, of the great 121ionization produced by the rays between the plates, any charge given to one of them is almost instantly dissipated. In many cases, the plate does become charged to a definite positive or negative potential depending on the metal, but this is due to the contact difference of potential between the plates, and would be produced whether the rays were charged or not. The ionization of the gas is greatly diminished by placing over the active material a metal screen which absorbs the α rays, but allows the β rays to pass through with little absorption.
The rapid loss of any charge communicated to the top plate can be very much reduced, either by diminishing the pressure of the gas surrounding it or by enclosing the plate with suitable insulators. In their experiments to determine the amount of charge carried by the radium rays, M. and Mme Curie[120] used the second method.
A metal disc MM (Fig. 26) is connected with an electrometer by the wire T. The disc and wire are completely surrounded by insulating matter ii. The whole is surrounded by a metal envelope EEEE connected with earth. On the lower side of the disc, the insulator and the metallic covering are very thin. This side is exposed to the rays of the radium R placed in a depression in a lead plate AA.
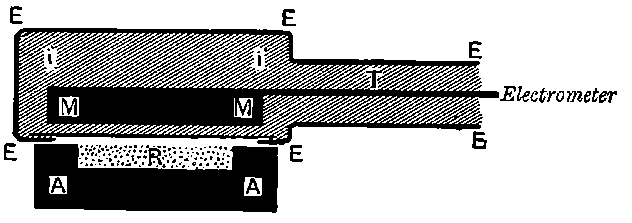
Fig. 26.
The rays of the radium pass through the metal cover and insulator with little absorption, but they are completely absorbed by the disc MM. It was observed that the disc received a negative charge which increased uniformly with the time, showing that the rays carry with them a negative charge. The current observed was very small. With an active preparation of radium[121], forming 122a layer 2·5 sq. cms. in area and 2 mms. thick, a current of the order of 10-11 amperes was observed after the rays had traversed a layer of aluminium ·01 mm. thick and a layer of ebonite ·3 mm. thick. The current was the same with discs of lead, copper, and zinc, and also when the ebonite was replaced by paraffin.
Curie also observed in another experiment of a similar character that the radium itself acquired a positive charge. This necessarily follows if the rays carry with them a negative charge. If the β rays alone carried with them a charge, a pellet of radium, if perfectly insulated, and surrounded by a non-conducting medium, would in the course of time be raised to a high positive potential. Since, however, the α rays carry with them a charge opposite in sign to the β rays, the ratio of the charge carried off by the two types of rays must be determined, before it can be settled whether the radium would acquire a positive or a negative charge. If, however, the radium is placed in an insulated metal vessel of a thickness sufficient to absorb all the α rays, but not too thick to allow most of the β rays to escape, the vessel will acquire a positive charge in a vacuum.
An interesting experimental result bearing upon this point has been described by Dorn[122]. A small quantity of radium was placed in a sealed glass tube and left for several months. On opening the tube with a file, a bright electric spark was observed at the moment of fracture, showing that there was a large difference of potential between the inside of the tube and the earth.
In this case the α rays were absorbed in the walls of the tube, but a large proportion of the β rays escaped. The inside of the tube thus became charged, in the course of time, to a high positive potential; a steady state would be reached when the rate of escape of negative electricity was balanced by the leakage of positive electricity through the walls of the tube. The external surface of the glass would be always practically at zero potential, on account of the ionization of the air around it.
Strutt[123] has recently described a simple and striking experiment to illustrate still more clearly that a radium preparation acquires a positive charge, if it is enclosed in an envelope thick enough to 123absorb all the α particles, but thin enough to allow most of the β particles to escape. The experimental arrangement is clearly seen in Fig. 27. A sealed tube AA containing the radium, was attached at one end to a pair of thin gold leaves in metallic connection with the radium, and was insulated inside a larger tube by means of a quartz rod B. The inner surface of the tube was coated with tinfoil EE connected to earth. The glass surface of AA was made conducting by a thin coating of phosphoric acid. The air in the outer tube was exhausted as completely as possible by means of a mercury pump, in order to reduce the ionization in the gas, and consequently the loss of any charge gained by the gold leaves. After an interval of 20 hours, the gold leaves were observed to diverge to their full extent, indicating that they had acquired a large positive charge. In this experiment Strutt used ½ gram of radiferous barium of activity only 100 times that of uranium.
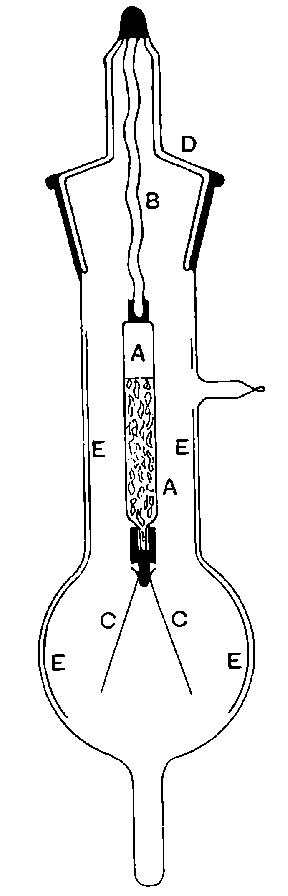
Fig. 27.
If the tube is filled with 30 mgrs. of pure radium bromide, the leaves diverge to their full extent in the course of about a minute. If it is arranged that the gold leaf, at a certain angle of divergence, comes in contact with a piece of metal connected with earth, the apparatus can be made to work automatically. The leaf diverges, touches the metal, and at once collapses, and this periodic movement of the leaf will continue, if not indefinitely, at any rate as long as the radium lasts. This “radium clock” should work at a sensibly uniform rate for many years, but, from evidence considered later (Section 261), there is reason to believe that the number of β particles emitted would decrease exponentially with the time, falling to half value in about 1200 years. The period of movement of the leaf should thus gradually increase with the time, and ultimately the effect would become too small to observe.
124The action of this radium clock is the nearest approach to an apparent perpetual motion that has so far been observed.
A determination of the amount of the charge carried off by the β rays of radium has been made by Wien[124]. A small quantity of radium, placed in a sealed platinum vessel, was hung by an insulating thread inside a glass cylinder, which was exhausted to a low pressure. A connection between the platinum vessel and an electrode sealed on to the external glass cylinder could be made, when required, by tilting the tube. Wien found that in a good vacuum the platinum vessel became charged to about 100 volts. The rate of escape of negative electricity from the platinum vessel containing 4 milligrams of radium bromide corresponded to 2·91 × 10-12 amperes. If the charge on each particle is taken as 1·1 × 10-20 electromagnetic units, this corresponds to an escape of 2·66 × 107 particles per second. From 1 gram of radium bromide the corresponding number would be 6·6 × 109 per second. Since some of the β rays are absorbed in their passage through the walls of the containing vessel and through the radium itself, the actual number projected per second from 1 gram of radium bromide must be greater than the above value. This has been found by the writer to be the case. The method employed reduced the absorption of the β rays to a minimum, and the total number emitted per second by 1 gram of radium bromide in radio-active equilibrium was found to be 4·1 × 1010, or about six times the number found by Wien. A detailed account of the method employed cannot be given with advantage at this stage, but will be found later in Section 253.
81. Determination of e/m. We have seen (Section 50) that, in their passage between the plates of a condenser, the cathode rays are deflected towards the positive plate. Shortly after the discovery of the magnetic deviation of the β rays from radium, Dorn[125] and Becquerel[126] showed that they also were deflected by an electric field.
By observing separately the amount of the electric and magnetic deviation, Becquerel was able to determine the ratio of e/m and the velocity of the projected particles. Two rectangular copper 125plates, 3·45 cms. high and 1 cm. apart, were placed in a vertical plane and insulated on paraffin blocks. One plate was charged to a high potential by means of an influence machine, and the other was connected with earth. The active matter was placed in a narrow groove cut in a lead plate parallel to the copper plates and placed midway between them. The photographic plate, enveloped in black paper, was placed horizontally above the plate containing the active substance. The large and diffuse pencil of rays thus obtained was deflected by the electric field, but the deviation amounted to only a few millimetres and was difficult to measure. The method finally adopted was to place vertically above the active matter a thin screen of mica, which cut the field into two equal parts. Thus, in the absence of an electric field, a narrow rectangular shadow was produced on the plate.
When the electric field was applied, the rays were deflected and a part of the pencil of rays was stopped by the mica screen. A shadow was thus cast on the plate which showed the direction of deviation and corresponded to the least deviable rays which gave an impression through the black paper.
If a particle of mass m, charge e, and velocity u, is projected normally to an electric field of strength X, the acceleration α is in the direction of the field, and is given by
Since the particle moves with a constant acceleration parallel to the field, the path of the particle is the same as that of a body projected horizontally from a height with a constant velocity and acted on by gravity. The path of the particle is thus a parabola, whose axis is parallel to the field and whose apex is at the point where the particle enters the electric field. The linear deviation d1 of the ray parallel to the field after traversing a distance l is given by
On leaving the electric field, the particle travels in the direction of the tangent to the path at that point. If θ is the angular deviation of the path at that point
126The photographic plate was at a distance h above the extremity of the field. Thus the particles struck the plate at a distance d2 from the original path given by
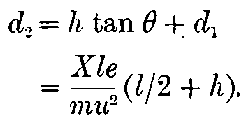
In the experimental arrangement the values were
If the radius R of curvature of the path of the same rays is observed in a magnetic field of strength H perpendicular to the rays,
Combining these two equations we get
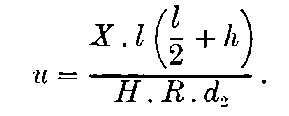
A difficulty arose in identifying the part of the complex pencil of rays for which the electric and magnetic deviations were determined. Becquerel estimated that the value of HR for the rays deflected by the electric field was about 1600 C.G.S. units. Thus
Thus these rays had a velocity more than half the velocity of light, and an apparent mass about the same as the cathode ray particles, i.e. about ¹⁄₁₀₀₀ of the mass of the hydrogen atom. The β ray is therefore analogous in all respects to the cathode ray, except that it differs in velocity. In a vacuum tube the cathode rays generally have a velocity of about 2 × 109 cms. per sec. In special tubes with strong fields the velocity may be increased to about 1010 cms. per sec. These β particles, then, behave like isolated units of negative electricity, identical with the electrons set free by an electric discharge in a vacuum tube. The electrons projected 127from radium have velocities varying from about 0·2V to at least 0·96V, where V is the velocity of light, and thus have an average speed considerably greater than that of the electrons produced in a vacuum tube. These moving electrons are able to pass through much greater thicknesses of matter before they are absorbed than the slower electrons produced in a vacuum tube, but the difference is one merely of degree and not of kind. Since electrons are continuously and spontaneously expelled from radium with enormous velocities, they must acquire their energy of motion from the matter itself. It is difficult to avoid the conclusion, that this velocity has not been suddenly impressed on the electron. Such a sudden gain of velocity would mean an immense and sudden concentration of energy on a small particle, and it is more probable that the electron before its expulsion has been in rapid orbital or oscillatory motion in the atom, and, by some means, suddenly escapes from its orbit. According to this view, the energy of the electron is not suddenly created but is only made obvious by its escape from the system to which it belongs.
82. Variation of e/m with the velocity of the electron. The fact that radium throws off electrons with rates of speed varying from ⅕ to ⁹⁄₁₀ the velocity of light has been utilised by Kaufmann[127] to examine whether the ratio e/m of the electrons varies with the speed. We have seen (Section 48) that, according to the electromagnetic theory, a charge of electricity in motion behaves as if it had apparent mass. For small speeds, this additional electrical mass is equal to
where a is the radius of the body, but it increases rapidly as the speed of light is approached. It is very important to settle whether the mass of the electron is due partly to mechanical and partly to electrical mass, or whether it can be explained by virtue of electricity in motion independently of the usual conception of mass.
Slightly different formulae expressing the variation of mass with speed have been developed by J. J. Thomson, Heaviside, and Searle. To interpret his results Kaufmann used a formula developed by M.
128Abraham[128].
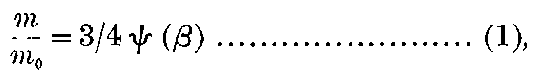
where
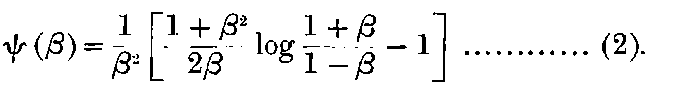
The experimental method employed to determine e/m and u is similar to the method of crossed spectra. Some strongly active radium was placed at the bottom of a brass box. The rays from this passed between two brass plates insulated and about 1·2 mm. apart. These rays fell on a platinum diaphragm, containing a small tube about 0·2 mm. in diameter, which allowed a narrow bundle of rays to pass. The rays then struck a photographic plate enveloped in a thin layer of aluminium.
In the experiments the diaphragm was about 2 cms. from the active material and at the same distance from the photographic plate. When the whole apparatus was placed in a vacuum, a P.D. of from 2000 to 5000 volts could be applied between the plates without a spark. The rays were deflected in their passage through the electric field, and produced what may be termed an electric spectrum on the plate.
Fig. 28.
If a magnetic field is superimposed parallel to the electric field by means of an electromagnet, a magnetic spectrum is obtained perpendicular to the electric spectrum. The combination of the two spectra gives rise to a curved line on the plate. The double trace obtained on the photographic plate with reversal of the magnetic field is shown in Fig. 28. Disregarding some small corrections, it can readily be shown that if y and z are the electric and magnetic deviations respectively,
129From these two equations, combined with (1), we obtain
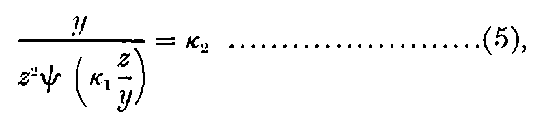
where κ, κ1, κ2 are constants.
Equation (5) gives the curve that should be obtained on the plate according to the electromagnetic theory. This is compared by trial with the actual curve obtained on the plate.
In this way Kaufmann[129] found that the value of e/m decreased with the speed, showing that, assuming the charge constant, the mass of the electron increased with the speed.
The following numbers give some of the preliminary results obtained by this method.
| Velocity of electron | e/m |
|---|---|
| 2·36 × 1010 cms. per sec. | 1·31 × 107 |
| 2·48 „ „ | 1·17 × 107 |
| 2·59 „ „ | 0·97 × 107 |
| 2·72 „ „ | 0·77 × 107 |
| 2·85 „ „ | 0·63 × 107 |
For the cathode rays S. Simon[130] obtained a value for e/m of 1·86 × 107 for an average speed of about 7 × 109 cms. per second.
In a later paper[131] with some very active radium, more satisfactory photographs were obtained, which allowed of accurate measurement. The given equation of the curve was found to agree satisfactorily with experiment.
The table given below, deduced from the results given by Kaufmann, shows the agreement between the theoretical and experimental values, u being the velocity of the electron and V that of light.
The average percentage error between the observed and calculated value is thus not much more than one per cent. It is 130remarkable how nearly the velocity of the electron has to approach the velocity of light before the value of m/m₀ becomes large. This is shown in the following table which gives the calculated values of m/m₀ for different velocities of the electron.
| Value of u/V | Observed value of m/m₀ | Percentage difference from theoretical values |
|---|---|---|
| Small | 1 | |
| ·732 | 1·34 | -1·5 % |
| ·752 | 1·37 | -0·9 „ |
| ·777 | 1·42 | -0·6 „ |
| ·801 | 1·47 | +0·5 „ |
| ·830 | 1·545 | +0·5 „ |
| ·860 | 1·65 | 0 „ |
| ·883 | 1·73 | +2·8 „ |
| ·933 | 2·05 | -7·8 „ ? |
| ·949 | 2·145 | -1·2 „ |
| ·963 | 2·42 | +0·4 „ |
| Value of u/V | small | ·1 | ·5 | ·9 | ·99 | ·999 | ·9999 | ·999999 |
| Calculated value m/m₀ | 1·00 | 1·015 | 1·12 | 1·81 | 3·28 | 4·96 | 6·68 | 10·1 |
Thus for velocities varying from 0 to ⅒ the velocity of light, the mass of the electron is practically constant. The increase of mass becomes appreciable at about half the velocity of light, and increases steadily as the velocity of light is approached. Theoretically the mass becomes infinite at the velocity of light, but even when the velocity of the electron only differs from that of light by one part in a million, its mass is only 10 times the value for slow speeds.
The above results are therefore in agreement with the view that the mass of the electron is altogether electrical in origin and can be explained purely by electricity in motion. The value of e/m₀, for slow speeds, deduced from the results was 1·84 × 107, which is in very close agreement with the value obtained by Simon for the cathode rays, viz. 1·86 × 107.
131If the electricity carried by the electron is supposed to be distributed uniformly over a sphere of radius a, for speeds slow compared with the velocity of light, the apparent mass
Therefore
Taking the value of e as 1·13 × 10-20, a is 1·4 × 10-13 cms.
Thus the diameter of an electron is minute compared with the diameter of an atom.
83. Distribution of velocity amongst the β particles. Some interesting experiments have been recently made by Paschen[132] to determine the relative number of β particles which are expelled from radium at the different speeds. The experimental arrangement is shown in Fig. 29.
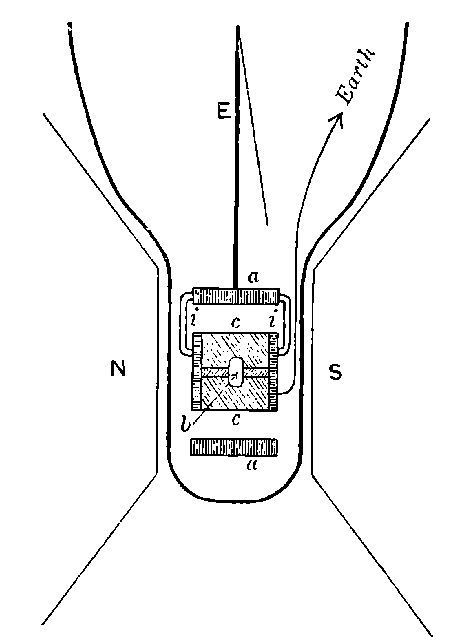
Fig. 29.
A small thin silvered glass tube b, containing 15 mgrs. of radium bromide, was placed in the axis of a number of lead vanes arranged round a cylinder of diameter 2 cms. and length 2·2 cms. 132When no magnetic field was acting, the β particles from the radium passed through the openings and were absorbed in an outer concentric cylinder aa of lead of inner diameter 3·7 cms. and of thickness 5·5 mms. This outer cylinder was rigidly connected to the inner cylinder cc by quartz rods ii, which also served to insulate it. The cylinder c and the radium were connected with earth. A gold-leaf electroscope E was attached to a, and the whole apparatus was enclosed in a glass vessel which was exhausted to a low vacuum by means of a mercury pump. The glass vessel was placed in the uniform field of a large electromagnet, so that the axis of the lead cylinder was parallel to the lines of force.
The outer cylinder gains a negative charge on account of the particles which are absorbed in it. This negative charge, which is indicated by the movement of the gold-leaf, tends to be dissipated by the small ionization produced in the residual gas by the passage of the β rays. This action of the gas can be eliminated by observing the rate of movement of the gold leaf when charged alternately to an initial positive and negative potential. The mean of the two rates is proportional to the number of β particles which give up their charge to the lead cylinder. This is evidently the case, since, when the charge is positive, the ionization of the gas assists the rate of movement of the gold-leaf, and, when negative, diminishes it to an equal extent.
When a magnetic field is applied, each of the particles describes a curved path, whose radius of curvature depends on the velocity of the particle. For weak fields, only the particles of smallest velocity will be deflected sufficiently not to strike the outer cylinder, but, as the field is raised, the number will increase until finally all the β particles fail to reach the outer cylinder. The decrease of the charge communicated to the outer cylinder with the increase of the strength of the magnetic field is shown graphically in Fig. 30, Curve I.
The ordinates represent in arbitrary units the charge communicated to the lead cylinder per second, and thus serve as a measure of the number of β particles which reach the cylinder. Knowing the dimensions of the apparatus, and assuming the value e/m found by Kaufmann, the velocity of the particles which just fail to reach the lead cylinder can be deduced from any strength 133of the magnetic field. Curve II, Fig. 30 is the first differential of Curve I, and the ordinates represent the relative number of β particles which are projected at each velocity.
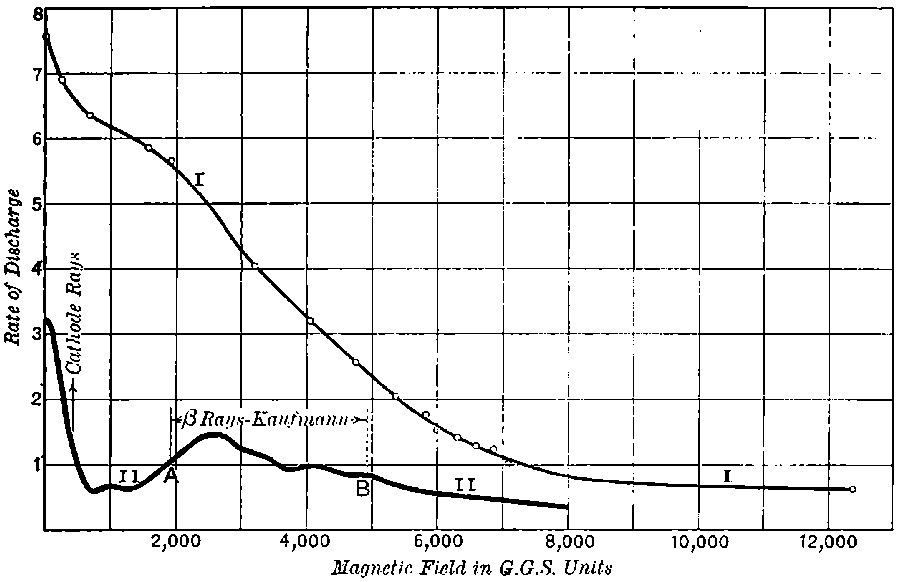
Fig. 30.
From the data given by Kaufmann (see section 82) Paschen deduced that the group of rays examined by the former, which had velocities lying between 2·12 × 1010 and 2·90 × 1010 cms. per second, corresponded to the group of rays between the points A and B, that is, to the group of rays which were completely deflected from the lead cylinder between the magnetic fields of strengths of 1875 and 4931 C.G.S. units. Since radium gives off β particles which require a field of strength over 7000 units to deflect them, Paschen concluded that β particles are expelled from radium with still greater velocities than the highest recorded by Kaufmann.
Paschen considered that the small charge observed in still higher fields was mainly due to the γ rays. The effect is small and is probably not due to an actual charge carried by the γ rays but to a secondary effect produced by them. This question will be discussed in more detail in section 112.
There is a group of low velocity β particles emitted by radium (see Fig. 30) which have about the same speed as the electrons 134set free in a vacuum tube. In consequence of their small velocity, these probably produce a large proportion of the ionization due to the β rays at short distances from the radium, for it will be shown (section 103) that the ionization produced by an electron per unit length of path steadily decreases with increase of its velocity above a small limiting value. This observation is confirmed by experiments on the absorption of the β rays in passing through matter.
In Paschen’s experiments, the glass tube containing the radium was ·5 mms. thick, so that a considerable proportion of the low velocity β particles must have been stopped by it. This is borne out by some later experiments of Seitz which will be described in section 85.
84. Absorption of the β rays by matter. The β particles produce ions in their passage through the gas and their energy of motion is consequently diminished. A similar action takes place also when the β rays pass through solid and liquid media, and the mechanism of absorption is probably similar in all cases. Some of the particles in their passage through matter are completely stopped, while others have their velocity reduced. In addition, there is a considerable scattering or diffuse reflection of the rays in traversing matter. The amount of this scattering depends upon the density of the substance and also upon the angle of incidence of the rays. This scattering of the rays will be discussed later in section 111.
There are two general methods of determining the absorption of the β rays. In the first method, the variation of the ionization current is observed in a testing vessel when the active matter is covered by screens differing in material and thickness. This ionization in the vessel depends upon two quantities, viz. the number of β particles which pass through the matter and also upon the number of ions produced by them per unit path. In the absence of any definite information in regard to the variation of ionization by the electron with its velocity, no very definite conclusions can be drawn from such experiments.
The advent of pure radium-bromide has made it possible to determine the actual number of electrons which are absorbed in their passage through a definite thickness of matter, by measuring 135the negative charge carried by the issuing rays. Experiments of this character have been made by Seitz and will be considered later.
These two methods of determining the absorption of β rays are quite distinct in principle, and it is not to be expected that the values of the coefficients of absorption obtained in the two cases should be the same. The whole question of the absorption of electrons by matter is very complicated, and the difficulty is still further increased by the complexity of the β rays emitted by the radio-active substances. Many of the results obtained by different methods, while pointing to the same general conclusion, are quantitatively in wide disagreement. Before any definite advance can be made to a better understanding of the mechanism of absorption, it will be necessary to determine the variation of the ionization with the speed of the electron over a very wide range. Some work has already been done in this direction but not between sufficiently wide limits.
We shall first consider the results obtained on the absorption of β rays by measuring the variation of the ionization current, when screens of different thickness are placed over the active substance. When the active matter is covered with aluminium foil of thickness ·1 mm., the current in a testing vessel such as is shown in Fig. 17, is due almost entirely to the β rays. If a uranium compound is used, it is found that the saturation current decreases with the thickness of matter traversed nearly according to an exponential law. Taking the saturation current as a measure of the intensity of the rays, the intensity I after passing through a thickness d of matter is given by
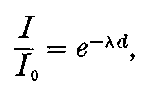
where λ is the constant of absorption of the rays and I₀ is the initial intensity. For uranium rays, the current is reduced to half its value after passing through about ·5 mm. of aluminium.
If a compound of thorium or radium is examined in the same way, it is found that the current does not decrease regularly 136according to the above equation. Results of this kind for radium rays have been given by Meyer and Schweidler[133]. The amount of absorption of the rays by a certain thickness of matter decreases with the thickness traversed. This is exactly opposite to what is observed for the α rays. This variation in the absorption is due to the fact that the β rays are made up of rays which vary greatly in penetrating power. The rays from uranium are fairly homogeneous in character, i.e. they consist of rays projected with about the same velocity. The rays from radium and thorium are complex, i.e. they consist of rays projected with a wide range of velocity and consequently with a wide range of penetrating power. The electrical examination of the deviable rays thus leads to the same results as their examination by the photographic method.
Results on the absorption of cathode rays have been given by Lenard[134], who has shown that the absorption of cathode rays is nearly proportional to the density of the absorbing matter, and is independent of its chemical state. If the deviable rays from active bodies are similar to cathode rays, a similar law of absorption is to be expected. Strutt[135], working with radium rays, has determined the law of absorption, and has found it roughly proportional to the density of matter over a range of densities varying from 0·041 for sulphur dioxide to 21·5 for platinum. In the case of mica and cardboard, the values of λ divided by the density were 3·94 and 3·84 respectively, while the value for platinum was 7·34. In order to deduce the absorption coefficient, he assumed that the radiation fell off according to an exponential law with the distance traversed. As the rays from radium are complex, we have seen that this is only approximately the case.
Since the β rays from uranium are fairly homogeneous, and are at the same time penetrating in character, they are more suitable for such a determination than the complex rays of radium. I have in consequence made some experiments with uranium rays to determine the dependence of absorption on the density. The results obtained are given in the following table, where λ is the coefficient of absorption.
| 137 | |||
| Substance | λ | Density | λ/Density |
|---|---|---|---|
| Glass | 14·0 | 2·45 | 5·7 |
| Mica | 14·2 | 2·78 | 5·1 |
| Ebonite | 6·5 | 1·14 | 5·7 |
| Wood | 2·16 | ·40 | 5·4 |
| Cardboard | 3·7 | ·70 | 5·3 |
| Iron | 44 | 7·8 | 5·6 |
| Aluminium | 14·0 | 2·60 | 5·4 |
| Copper | 60 | 8·6 | 7·0 |
| Silver | 75 | 10·5 | 7·1 |
| Lead | 122 | 11·5 | 10·8 |
| Tin | 96 | 7·3 | 13·2 |
It will be observed that the value of the absorption constant divided by the density is very nearly the same for such different substances as glass, mica, ebonite, wood, iron and aluminium. The divergences from the law are great, however, for the other metals examined, viz. copper, silver, lead and tin. In tin the value of λ divided by the density is 2·5 times its value for iron and aluminium. These differences show that a law for the absorption of the β rays depending only on the density does not hold for all substances. With an exception in the case of tin, the value of λ divided by the density for the metals increases in the same order as their atomic weights.
The absorption of the β rays by matter decreases very rapidly with increase of speed. For example, the absorption of cathode rays in Lenard’s experiment (loc. cit.) is about 500 times as great as for the uranium β rays. The velocity of the β rays of uranium was found by Becquerel to be about 1·6 × 1010 cms. per sec. The velocity of the cathode rays used in Lenard’s experiment was certainly not less than ⅒ of this, so that, for a decrease of speed of less than 10 times, the absorption has increased over 500 times.
85. Number of electrons stopped by matter. An account will now be given of the experiments made by Seitz[136], to determine 138the relative number of electrons which are stopped in their passage through different thicknesses of matter. The experimental arrangement is shown in Fig. 31.
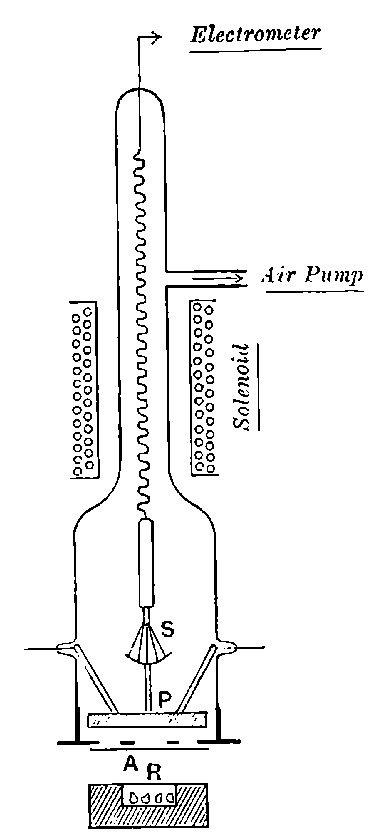
Fig. 31.
The radium was placed outside a glass vessel containing an insulated brass plate P, the connection of which with a wire leading to the electrometer could be made or broken by a simple electromagnetic device. The β rays from the radium R, after passing through openings in a brass plate A, covered with thin aluminium foil, were absorbed in the plate P. The glass vessel was exhausted, and the charge communicated to P by the β rays was measured by an electrometer.
In a good vacuum, the magnitude of the current observed is a measure of the number of β particles absorbed by the upper plate[137]. The following table shows the results obtained when different thicknesses of tin foil were placed over the radium. The second table gives the ratio I/I₀ where I₀ is the rate of discharge observed before the absorbing screen is introduced. The mean value of the absorption constant λ was deduced from the equation
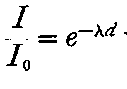
where d is the thickness of matter traversed.
The values included in the brackets have not the same accuracy as the others. There is thus a wide difference in penetrating power of the β particles emitted from radium, and some of them are very readily absorbed.
139When a lead screen 3 mms. thick was placed over the radium—a thickness sufficient to absorb all the readily deflectable β rays—a small negative charge was still given to the plate, corresponding to ·29 per cent. of the maximum. This is a very much smaller value than was observed by Paschen (see Fig. 30).
| Thickness of Tin in mms. | I/I₀ | λ |
|---|---|---|
| 0·00834 | ·869 | 175 |
| 0·0166 | ·802 | 132·5 |
| 0·0421 | ·653 | 101·5 |
| 0·0818 | ·466 | 93·5 |
| 0·124 | ·359 | 82·5 |
| 0·166 | ·289 | 74·9 |
| 0·205 | ·230 | 71·5 |
| 0·270 | ·170 | 65·4 |
| 0·518 | ·065 } | 53} |
| 0·789 | ·031 } | 44} |
| 1·585 | ·0059} | 32} |
| 2·16 | ·0043} | 25} |
This difference may, in part, be due to the fact that, in Paschen’s experiments, a large proportion of the slow velocity electrons were absorbed in the glass tube of ·5 mm. thickness containing the radium.
Seitz also determined the relative thickness, compared with tin, of different substances which reduced the negative charge communicated to P by a definite amount. A few of the numbers are given below, and expressed in terms of tin as unity.
| Substance | Thickness Tin = 1 |
|---|---|
| Lead | ·745 |
| Gold | ·83 |
| Platinum | ·84 |
| Silver | 1 |
| Steel | 1·29 |
| Aluminium | 1·56 |
| Water | 1·66 |
| Paraffin | 1·69 |
The thickness required to stop a given proportion of the β rays thus decreases with the density, but not nearly so fast as the 140density increases. These results are difficult to reconcile with the density-law of absorption found by Lenard from the cathode rays, or with the results of the ionization method already considered. A further experimental examination of the whole question is very much to be desired.
86. Variation of the amount of radiation with the thickness of the layer of radiating material. The radiations are sent out equally from all portions of the active mass, but the ionization of the gas which is measured is due only to the radiations which escape into the air. The depth from which the radiations can reach the surface depends on the absorption of the radiation by the active matter itself.
Let λ be the absorption constant of the homogeneous radiation by the active material. It can readily be shown that the intensity I of the rays issuing from a layer of active matter, of thickness d, is given by
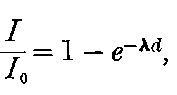
where I₀ is the intensity at the surface due to a very thick layer.
This equation has been confirmed experimentally by observing the current due to the β rays for different thicknesses of uranium oxide. In this case I = (½)I₀ for a thickness of oxide corresponding to ·11 gr. per sq. cm. This gives a value of λ divided by density of 6·3. This is a value slightly greater than that observed for the absorption of the same rays in aluminium. Such a result shows clearly that the substance which gives rise to the β rays does not absorb them to a much greater extent than does ordinary matter of the same density.
The value of λ will vary, not only for the different active substances, but also for the different compounds of the same substance.
87. The α rays. The magnetic deviation of the β rays was discovered towards the end of 1899, at a comparatively early stage in the history of radio-activity, but three years elapsed before the true character of the α rays was disclosed. It was natural that great prominence should have been given in the early stages of the subject to the β rays, on account of their great penetrating power and marked action in causing phosphorescence in many substances. The α rays were, in comparison, very little studied, and their importance was not generally recognized. It will, however, be shown that the α rays play a far more important part in radio-active processes than the β rays, and that the greater portion of the energy emitted in the form of ionizing radiations is due to them.
88. The nature of the α rays. The nature of the α rays was difficult to determine, for a magnetic field sufficient to cause considerable deviation of the β rays produced no appreciable effect on the α rays. It was suggested by several observers that they were, in reality, secondary rays set up by the β or cathode rays in the active matter from which they were produced. Such a view, however, failed to explain the radio-activity of polonium, which gave out α rays only. Later work also showed that the matter, which gave rise to the β rays from uranium, could be chemically separated from the uranium, while the intensity of the α rays was unaffected. These and other results show that the α and β rays are produced quite independently of one another. The view that they are an easily absorbed type of Röntgen rays fails to explain a characteristic property of the α rays, viz. that the absorption of the rays in a given thickness of matter, determined by the electrical method, increases with the thickness of matter previously traversed. It does not seem probable that such an effect could be produced by a radiation like X rays, but the result is to be expected if the rays consist of projected bodies, which fail to 142ionize the gas when their velocity is reduced below a certain value. From observations of the relative ionization produced in gases by the α and β rays, Strutt[138] suggested in 1901 that the α rays might consist of positively charged bodies projected with great velocity. Sir William Crookes[139], in 1902, advanced the same hypothesis. From a study of the α rays of polonium Mme. Curie[140] in 1900 suggested the probability that these rays consisted of bodies, projected with great velocity, which lost their energy by passing through matter.
The writer was led independently to the same view by a mass of indirect evidence which received an explanation only on the hypothesis that the rays consisted of matter projected with great velocity. Preliminary experiments with radium of activity 1000 showed that it was very difficult to determine the magnetic deviation of the α rays. When the rays were passed through slits sufficiently narrow to enable a minute deviation of the rays to be detected, the ionizing effect of the issuing rays was too small to be measured with certainty. It was not until radium of activity 19,000 was obtained that it was possible to detect the deviation of these rays in an intense magnetic field. How small the magnetic deviation is may be judged from the fact that the α rays, projected at right angles to a magnetic field of 10,000 C.G.S. units, describe the arc of a circle of about 39 cms. radius, while under the same conditions the cathode rays produced in a vacuum tube would describe a circle of about ·01 cm. radius. It is therefore not surprising that the α rays were for some time thought to be non-deviable in a magnetic field.
89. Magnetic deviation of the α rays. The general method employed[141] to detect the magnetic deviation of the α rays was to allow the rays to pass through narrow slits and to observe whether the rate of discharge of an electroscope, due to the issuing rays, was altered by the application of a strong magnetic field. Fig. 32 shows the general arrangement of the experiment. The 143rays from a thin layer of radium of activity 19,000 passed upwards through a number of narrow slits G, in parallel, and then through a thin layer of aluminium foil, ·00034 cm. thick, into the testing vessel V. The ionization produced by the rays in the testing vessel was measured by the rate of movement of the leaves of a gold-leaf electroscope B. The gold-leaf system was insulated inside the vessel by a sulphur bead C, and could be charged by means of a movable wire D, which was afterwards earthed. The rate of movement of the gold-leaf was observed through small mica windows in the testing vessel by means of a microscope provided with a micrometer eye-piece.
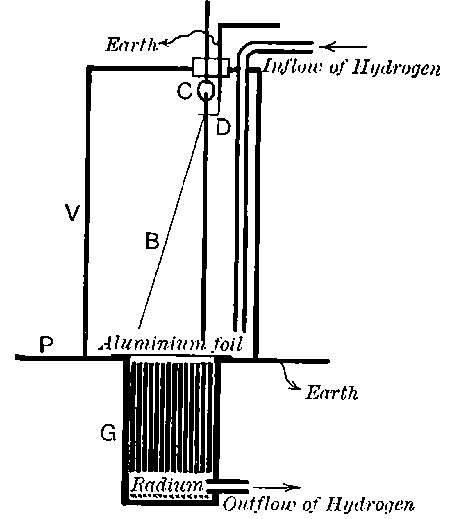
Fig. 32.
In order to increase the ionization in the testing vessel, the rays passed through 20 to 25 slits of equal width, placed side by side. This was arranged by cutting grooves at regular intervals in side-plates into which brass plates were slipped. The width of the slit varied in different experiments between ·042 cm. and ·1 cm. The magnetic field was applied perpendicular to the plane of the paper, and parallel to the plane of the slits. The rays are thus deflected in a direction perpendicular to the plane of the slits and a very small amount of deviation is sufficient to cause the rays to impinge on the sides of the plate where they are absorbed.
The testing vessel and system of plates were waxed to a lead plate P so that the rays entered the vessel V only through the 144aluminium foil. It is necessary in these experiments to have a steady stream of gas passing downwards between the plates in order to prevent the diffusion of the emanation from the radium upwards into the testing vessel. The presence in the testing vessel of a small amount of this emanation, which is always given out by radium, would produce great ionization and completely mask the effect to be observed. For this purpose, a steady current of dry electrolytic hydrogen of about 2 c.c. per second was passed into the testing vessel; it then streamed through the porous aluminium foil, and passed between the plates carrying the emanation with it away from the apparatus. The use of a stream of hydrogen instead of air greatly simplifies the experiment, for it increases the ionization current due to the α rays in the testing vessel, and at the same time greatly diminishes that due to the β and γ rays. This is caused by the fact that the α rays are much more readily absorbed in air than in hydrogen, while the rate of production of ions due to the β and γ rays is much less in hydrogen than in air. The intensity of the α rays after passing between the plates is consequently greater when hydrogen is used; and since the rays pass through a sufficient distance of hydrogen in the testing vessel to be largely absorbed, the total amount of ionization produced by them is greater with hydrogen than with air.
The following is an example of an observation on the magnetic deviation:—
| Rate of discharge of electroscope in volts per minute | |
|---|---|
| (1) Without magnetic field | 8·33 |
| (2) With magnetic field | 1·72 |
| (3) Radium covered with thin layer of mica to absorb all α rays | 0·93 |
| (4) Radium covered with mica and magnetic field applied | 0·92 |
145The mica plate, ·01 cm. thick, was of sufficient thickness to absorb completely all the α rays, while it allowed the β rays and γ rays to pass through without appreciable absorption. The difference between (1) and (3), 7·40 volts per minute, gives the rate of discharge due to the α rays alone; the difference between (2) and (3), 0·79 volts per minute, that due to the α rays not deviated by the magnetic field employed.
The amount of α rays not deviated by the field is thus about 11% of the total. The small difference between (3) and (4) measures the small ionization due to the β rays, for they would be completely deviated by the magnetic field; (4) comprises the effect of the γ rays together with the natural leak of the electroscope in hydrogen.
In this experiment there was a good deal of stray magnetic field acting on the rays before they reached the pole-pieces. The diminution of the rate of discharge due to the α rays was found to be proportional to the strength of field between the pole-pieces. With a more powerful magnetic field, the whole of the α rays were deviated, showing that they consisted entirely of projected charged particles.
In order to determine the direction of deviation of the rays, the rays were passed through slits one mm. in width, each of which was half covered with a brass strip. The diminution of the rate of discharge in the testing vessel for a given magnetic field in such a case depends upon the direction of the field. In this way it was found that the rays were deviated in the opposite sense to the cathode rays. Since the latter consist of negatively charged particles, the α rays must consist of positively charged particles.
These results were soon after confirmed by Becquerel[142], by the photographic method, which is very well adapted to determine the character of the path of the rays acted on by a magnetic field. The radium was placed in a linear groove cut in a small block of lead. Above this source, at a distance of about 1 centimetre, was placed a metallic screen, formed of two plates, leaving between them a narrow opening parallel to the groove. Above this was placed the photographic plate. The whole apparatus was placed in a strong magnetic field parallel to the groove. The strength of the 146magnetic field was sufficient to deflect the β rays completely away from the plate. When the plate was parallel to the opening, there was produced on it an impression, due to the α rays alone, which became more and more diffuse as the distance from the opening increased. This distance should not exceed 1 or 2 centimetres on account of the absorption of the rays in air. If, during the exposure, the magnetic field is reversed for equal lengths of time, on developing the plate two images of the α rays are observed which are deflected in opposite directions. This deviation, even in a strong field, is small though quite appreciable and is opposite in sense to the deviation observed for the β or cathodic rays from the same material.
M. Becquerel[143], by the same method, found that the α rays from polonium were deviated in the same direction as the α rays from radium; and thus that they also consist of projected positive bodies. In both cases, the photographic impressions were sharply marked and did not show the same diffusion which always appears in photographs of the β rays.
90. Electrostatic deviation of the α rays. If the rays are charged bodies, they should be deflected in passing through a strong electric field. This was found by the writer to be the case, but the electric deviation is still more difficult to detect than the magnetic deviation, as the intensity of the electric field must of necessity be less than that required to produce a spark in the presence of radium. The apparatus was similar to that employed for the magnetic deviation (Fig. 32) with this exception, that the brass sides which held the plates in position, were replaced by ebonite. Alternate plates were connected together and charged to a high potential by means of a battery of small accumulators. The discharge in the electroscope, due to the α rays, was found to be diminished by application of the electric field. With plates ·055 cm. apart and 4·5 cms. high, the diminution was only 7% with a P.D. of 600 volts between the slits. With a special arrangement of plates, with slits only ·01 cm. apart, the discharge was diminished about 45% with an electric field corresponding to 10,000 volts per cm.
147 91. Determination of the constants of the rays. If the deviation of the rays in both an electric and magnetic field is known, the values of the velocity of the rays, and the ratio e/m of the charge of the particle to its mass can be determined by the method, first used by J. J. Thomson for the cathode rays, which is described in section 50. From the equations of a moving charged body, the radius of curvature ρ of the path of the rays in a magnetic field of strength H perpendicular to the path of the rays is given by
If the particle, after passing through a uniform magnetic field for a distance l1, is deviated through a small distance d1 from its original direction,
If the rays pass through a uniform electric field of strength X and length l2 with a deviation d2,
since Xe/m is the acceleration of the particle, at right angles to its direction, and l2/V is the time required to travel through the electric field.
From equations (1) and (2)
The values of V and e/m are thus completely determined from the combined results of the electric and magnetic deviation. It was found that
On account of the difficulty of obtaining a large electrostatic deviation, these values are only approximate in character.
148The results on the magnetic and electric deviation of the α rays of radium have been confirmed by Des Coudres[144], by the photographic method. Some pure radium bromide was used as a source of radiation. The whole apparatus was enclosed in a vessel which was exhausted to a low vacuum. In this way, not only was he able to determine the photographic action of the rays at a much greater distance from the source, but he was also able to apply a stronger electric field without the passage of a spark. He found values of the constants given by
These values are in very good agreement with the numbers found by the electric method. The α rays from radium are complex, and probably consist of a stream of positively charged bodies projected at velocities lying between certain limits. The amount of deviation of the particles in a magnetic field will thus differ according to the velocity of the particle. The photographic results of Becquerel seem to indicate that the velocity of the rays of radium can vary only within fairly narrow limits, since the trajectory of the rays in a magnetic field is sharply marked and not nearly as diffuse as in similar experiments with the β rays. The evidence, however, discussed in the following section, shows that the velocities of the α particles from a thick layer of radium vary over a considerable range.
92. Becquerel[145] has examined the amount of magnetic deviation of the α rays at different distances from the source of the rays in a very simple way. A narrow vertical pencil of the rays, after its passage through a narrow slit, fell on a photographic plate, which was inclined at a small angle to the vertical and had its lower edge perpendicular to the slit. The trajectory of the rays is shown by a fine line traced on the plate. If a strong magnetic field is applied parallel to the slit, the trajectory of the rays is displaced to the right or left according to the direction of the field. If equal times of exposure are given for the magnetic field 149in the two directions, on developing the plate two fine diverging lines are found traced on the plate. The distance between these lines at any point is a measure of twice the average deviation at that point, corresponding to the value of the magnetic field. By measuring the distance between the trajectories at various points, Becquerel found that the radius of curvature of the path of the rays increased with the distance from the slit. The product Hρ of the strength of the field and the radius of curvature of the path of the rays is shown in the following table.
| Distance in mms. from the slit | Hρ |
|---|---|
| 1 | 2·91 × 105 |
| 3 | 2·99 „ |
| 5 | 3·06 „ |
| 7 | 3·15 „ |
| 8 | 3·27 „ |
| 9 | 3·41 „ |
The writer (loc. cit.) showed that the maximum value of Hρ for complete deviation of the α rays was 390,000. The results are thus in good agreement. Since
these results show that the values either of V or of e/m for the projected particles vary at different distances from the source. Becquerel considered that the rays were homogeneous, and, in order to explain the results, has suggested that the charge on the projected particles may gradually decrease with the distance traversed, so that the radius of curvature of the path steadily increases with the distance from the source. It, however, seems more probable that the rays consist of particles projected with different velocities, and that the slower particles are more quickly absorbed in the gas. In consequence of this, only the swifter particles are present some distance from the source.
This conclusion is borne out by some recent experiments of Bragg and Kleeman[146] on the nature of the absorption of α particles by matter, which are discussed in more detail in sections 103 and 104. They found that the α particles from a thick layer of radium are complex, and have a wide range of penetrating power and presumably of velocity. This is due to the fact that the α particles 150emitted from the radium come from different depths. Since their velocity is reduced in their transit through matter, a pencil of α rays will consist of particles which differ considerably in speed. Those which are just able to emerge from the radium will be absorbed in a very short depth of air, while those that come from the surface will be able to pass through several centimetres of air before they lose their power of ionizing the gas. Since the α particles have different velocities, they will be unequally deflected by the magnetic field, the slower moving particles describing a more curved path than the swifter ones. Consequently, the outer edge of the trace of the pencil of rays on the photographic plate, as obtained by Becquerel, will be the locus of the points where the photographic action of the α particles end. It was found that the α particles are most efficient as ionizers of the gas just before their power of ionizing ends. The loss of ionizing power of the α particles seems to be fairly abrupt, and, for particles of the same velocity, to occur always after traversing a definite distance in air. On the assumption that the photographic as well as the ionizing action is most intense just before the particles are stopped, and ceases fairly abruptly, Bragg has been able to account numerically for the measurements (see above table) recorded by Becquerel. Quite apart from the special assumptions required for such a quantitative comparison of theory with experiment, there can be little doubt that the increase of value of Hρ with distance can be satisfactorily explained as a consequence of the complex character of the pencil of rays[147].
Becquerel states that the amount of deviation, in a given magnetic field, was the same for the α rays of polonium and of radium. This shows that the value of
is the same for the α rays from the two substances. Since the α rays from polonium are far more readily absorbed than the α rays from radium, this result would indicate that the value of m/e is greater for the α particles of polonium than of radium. Further experimental evidence is required on this important point.
151 93. Charge carried by the α rays. We have seen that the negative charge carried by the β particles has been readily measured. Since there is reason to believe (section 229) that four α particles are expelled from radium for each β particle, it is to be expected that the positive charge carried by the α particles should be determined still more readily. All the initial experiments, however, made to detect this charge, gave negative results; and, before successful results were obtained, it was found necessary to eliminate some secondary actions, which at first completely masked the effects to be looked for.
In consequence of the importance of this question, a brief account will be given of the methods of measurement adopted and the special experimental difficulties which have arisen.
In the first place, it must be remembered that only a small fraction of the α rays, emitted from a layer of powdered radium bromide, escape into the surrounding gas. On account of the ease with which the α rays are stopped in their passage through matter, only those escape which are expelled from a superficial layer, and the rest are absorbed by the radium itself. On the other hand, a much larger proportion of the β rays escape, on account of their greater power of penetration. In the second place, the α particle is a far more efficient ionizer of the gas than the β particle, and, in consequence, if the charge carried by the α rays is to be determined by methods similar to those employed for the β rays (see section 80), the pressure of the gas surrounding the conductor to be charged must be very small in order to eliminate, as far as possible, the loss of charge resulting from the ionization of the residual gas by the α rays[148].
The experimental arrangement used by the writer is shown in Fig. 33.
A thin film of radium was obtained on a plate A by evaporation of a radium solution containing a known weight of radium bromide. Some hours after evaporation, the activity of the radium, measured by the α rays, is about 25 per cent. of its maximum value, and the β rays are almost completely absent. The activity measured by the α and β rays is then slowly regained, and recovers its original value after about a month’s interval (see 152chapter XI.). The experiments were made on the active plate when its activity was a minimum, in order to avoid complications due to the presence of β rays. The film of radium was so thin that only a very small fraction of the α rays was absorbed.
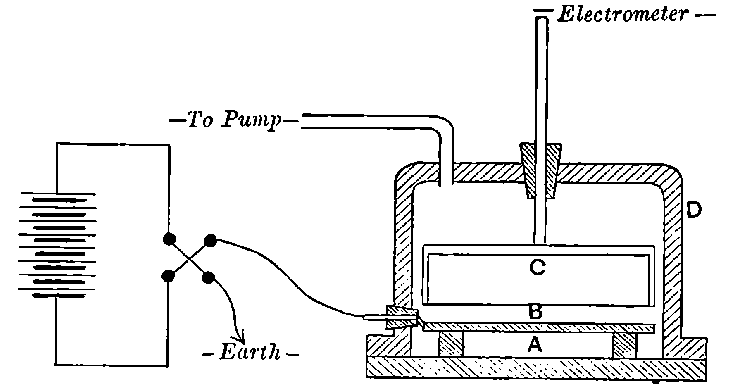
Fig. 33.
The active plate A was insulated in a metal vessel D, and was connected to one pole of the battery, the other pole being earthed. The upper electrode, which was insulated and connected with a Dolezalek electrometer, consisted of a rectangular copper vessel BC, the lower part of which was covered with a thin sheet of aluminium foil. The α rays passed through the foil, but were stopped by the copper sides of the vessel. This arrangement was found to reduce the secondary ionization produced at the surface of the upper plate. The outside vessel D could be connected with either A or B or with earth. By means of a mercury pump, the vessel was exhausted to a very low pressure. If the rays carry a positive charge, the current between the two plates measured by the electrometer should be greater when A is charged positively. No certain difference, however, between the currents in the two directions was observed, even when a very good vacuum was obtained. In some arrangements, it was found that the current was even greater when the lower plate was negative than when it was positive. An unexpected experimental result was also noticed. The current between the parallel plates at first diminished with the pressure, but soon reached a limiting value which was not altered however good a vacuum was produced. For example, in one experiment, the current between 153the two parallel plates, placed about 3 mms. apart, was initially 6·5 × 10-9 amperes and fell off directly as the pressure. The current reached a limiting value of about 6 × 10-12 amperes, or about ¹⁄₁₀₀₀ of the value at atmospheric pressure. The magnitude of this limiting current was not much altered if the air was replaced by hydrogen.
Experiments of a similar character have been made by Strutt[149] and J. J. Thomson[150]; using an active bismuth plate coated with radio-tellurium (polonium) after Marckwald’s method. This substance emits only α rays, and is thus especially suitable for experiments of this kind. Strutt employed the method used by him to show the charge carried by the β rays (Fig. 27). He found, however, that, even in the lowest possible vacuum, the electroscope rapidly lost its charge and at the same rate whether it was charged positively or negatively. This is in agreement with the results found by the writer with radium.
In the experiments of J. J. Thomson, the electroscope was attached to a metal disc placed 3 cms. from the plate of radio-tellurium. A very low vacuum was produced by Dewar’s method by absorbing the residual gas in cocoanut charcoal immersed in liquid air. When the electroscope was charged negatively, an extremely slow rate of leak was observed, but when charged positively the leak was about 100 times greater. This showed that the polonium gave out large quantities of negative electricity, but not enough positive to be detected. By placing the apparatus in a strong magnetic field, the negative particles were prevented from reaching the electroscope and the positive leak was stopped.
These results indicate that these negative particles are not projected with sufficient velocity to move against the repulsion exerted by the electrified body, and are bent by a magnetic field. There thus seems little doubt that a stream of negative particles (electrons) is projected from the active surface at a very slow speed. Such low velocity electrons are also projected from uranium and radium. It is probable that these electrons are 154a type of secondary radiation, set up at the surfaces on which the α rays fall. The particles would be extremely readily absorbed in the gas, and their presence would be difficult to detect except in low vacua. J. J. Thomson at first obtained no evidence that the α particles of polonium were charged; but in later experiments, where the plates were closer together, the electroscope indicated that the α rays did carry a positive charge.
In order to see whether the positive charge due to the α rays from radium could be detected when the slow moving ions were prevented from escaping by a magnetic field, I placed the apparatus of Fig. 33 between the pole-pieces of a large electromagnet, so that the magnetic field was parallel to the plane of the plates[151]. A very marked alteration was observed both on the magnitude of the positive and negative currents. In a good vacuum, the upper plate received a positive charge, independently of whether the lower plate was charged positively or negatively or was connected with earth. After the magnetic field had reached a certain value, a great increase in its strength had no appreciable effect on the magnitude of the current.
The following table illustrates the results obtained when the two plates were 3 mms. apart, and were both coated with thin aluminium foil.
| Potential of lower plate | Current in | arbitrary units | |
|---|---|---|---|
| Without magnetic field | With magnetic field | ||
| 0 | — | +·36 | |
| +2 volts | 2·0 | +·46} | |
| } | ·39 | ||
| -2 „ | 2·5 | +·33} | |
| +4 „ | 2·8 | +·47} | |
| } | ·41 | ||
| -4 „ | 3·5 | +·35} | |
| +8 „ | 3·1 | +·56} | |
| } | ·43 | ||
| -8 „ | 4·0 | +·31} | |
| +84 „ | 3·5 | +·77} | |
| } | ·50 | ||
| -84 „ | 5·2 | +·24} |
Let n be the number of α particles, carrying a charge e, which are absorbed in the upper plate. Let ι₀ be the current due to the slight ionization of the residual gas.
If only a small potential is applied to the lower plate, this current should be equal in magnitude but opposite in sign when 155the potential is reversed. Let ι1 be the charge per sec. communicated to the upper electrode when the lower plate is charged positively and ι2 the value when charged negatively. Then
Now in the third column of the above table it is seen that (ι1 + ι2)/2 has the values ·39, ·41, ·43 for 2, 4, and 8 volts respectively. The numbers are thus in fairly good agreement. Similar results were obtained when a brass plate was substituted for the upper electrode shown in the figure. Taking into consideration that the magnitude of ne is independent of the strength of the magnetic field above a certain small value, and the good agreement of the numbers obtained with variation of voltage, I think that there can be no doubt that the positive charge communicated to the upper electrode was carried by the α particles. This positive charge was not small, for using a weight of ·48 mgrs. radium bromide spread in a thin foil over an area of about 20 sq. cms., the charge communicated by the particles corresponded to a current 8·8 × 10-13 amperes, and, with the Dolezalek electrometer employed, it was necessary to add a capacity of ·0024 microfarads to the electrometer system.
In these experiments, the film of radium bromide was so thin, that only a very small percentage of the α particles was stopped by the radium itself. Assuming that each α particle carries the same charge as an ion, viz. 1·1 × 10-19 coulombs, and remembering that half of the α particles are absorbed in the lower plate, the total number N of α particles expelled per second from one gram of radium bromide (at its minimum activity) can be deduced. In two separate experiments where the amount of radium used was ·194 and ·484 mgrs. respectively, the values of N were in close agreement and equal to 3·6 × 1010. Now it will be shown later that in radium there are three other products in radio-active equilibrium, each of which probably gives out the same number of α particles as radium itself. If this is the case, the total number of α particles expelled per second from 1 gram of radium bromide in radio-active equilibrium is 4N or 1·44 × 1011. Assuming the 156composition of radium bromide as RaBr2, the number per second per gram of radium is 2·5 × 1010. This number will be found to be in very good agreement with that deduced from indirect data (chapter XIII.). The value of N is of great importance in determining the magnitude of various quantities in radio-active calculations.
94. Mass and energy of the α particle. It has been pointed out that the α rays from radium and polonium are analogous to the Canal rays of Goldstein, for both carry a positive charge and are difficult to deflect by a magnetic field. The experiments of Wien have shown that the velocity of projection of the canal rays varies with the gas in the tube and the intensity of the electric field applied, but it is generally about ⅒ of the velocity of the α particle from radium. The value of e/m is also variable, depending upon the gas in the tube.
It has been shown that for the α rays of radium
Now the value of e/m for the hydrogen atom, liberated in the electrolysis of water, is 104. Assuming the charge carried by the α particle to be the same as that carried by the hydrogen atom, the mass of the α particle is about twice that of the hydrogen atom. Taking into consideration the uncertainty attaching to the experimental value of e/m for the α particle, if the α particle consists of any known kind of matter, this result indicates that it consists either of projected helium or hydrogen. Further evidence on this important question is given in section 260.
The α rays from all the radio-active substances and their products, such as the radio-active emanations and the matter causing excited activity, possess the same general properties and do not vary very much in penetrating power. It is thus probable that in all cases the α rays from the different radio-active substances consist of positively charged bodies projected with great velocity. Since the rays from radium are made up in part of α rays from the emanation stored in the radium, and from the excited activity which it produces, the α rays from each of these products must consist of positively charged bodies; for it has been shown that all the α rays from radium are deviated in a strong magnetic field.
157The kinetic energy of each projected particle is enormous, compared with its mass. The kinetic energy of each α particle is
Taking the velocity of a rifle bullet as 105 cms. per second, it is seen that, mass for mass, the energy of motion of the α rays is 6 × 108 times as great as that of the rifle bullet. In this projection of bodies atomic in size with great velocity probably lies the principal cause of the heating effects produced by radium (chapter XII).
95. Atomic disintegration. The radio-activity of the radio-elements is an atomic and not a molecular property. The rate of emission of the radiations depends only on the amount of the element present and is independent of its combination with inactive substances. In addition, it will be shown later that the rate of emission is not affected by wide variations of temperature, or by the application of any known chemical or physical forces. Since the power of radiating is a property of the radio-atoms, and the radiations consist for the most part of positively and negatively charged masses projected with great velocity, it is necessary to suppose that the atoms of the radio-elements are undergoing disintegration, in the course of which parts of the atom escape from the atomic system. It seems very improbable that the α and β particles can suddenly acquire their enormous velocity of projection by the action of forces existing inside or outside the atom. For example, the α particle would have to travel from rest between two points differing in potential by 5·2 million volts in order to acquire the kinetic energy with which it escapes. Thus it seems probable that these particles are not set suddenly in motion, but that they escape from an atomic system in which they were already in rapid oscillatory or orbital motion. On this view, the energy is not communicated to the projected particles, but exists beforehand in the atoms from which they escape. The idea that the atom is a complicated structure consisting of charged parts in rapid oscillatory or orbital motion has been developed by J. J. Thomson, Larmor and Lorentz. Since the α particle is atomic in size, it is 158natural to suppose that the atoms of the radio-active elements consist not only of the electrons in motion, but also of positively charged particles whose mass is about the same as that of the hydrogen or helium atom.
It will be shown later that only a minute fraction of the atoms of the radio-element need break up per second in order to account for the radiations even of an enormously active element like radium. The question of the possible causes which lead to this atomic disintegration and the consequences which follow from it will be discussed later in chapter XIII.
96. Experiments with a zinc sulphide screen. A screen of Sidot’s hexagonal blend (phosphorescent crystalline zinc sulphide) lights up brightly under the action of the α rays of radium and polonium. If the surface of the screen is examined with a magnifying glass, the light from the screen is found not to be uniformly distributed but to consist of a number of scintillating points of light. No two flashes succeed one another at the same point, but they are scattered over the surface, coming and going rapidly without any movement of translation. This remarkable action of the radium and polonium rays on a zinc sulphide screen was discovered by Sir William Crookes[152], and independently by Elster and Geitel[153], who observed it with the rays given out from a wire which has been charged negatively either in the open air or in a vessel containing the emanation of thorium.
In order to show the scintillations of radium on the screen, Sir William Crookes has devised a simple apparatus which he has called the “Spinthariscope.” A small piece of metal, which has been dipped in a radium solution, is placed several millimetres away from a small zinc sulphide screen. This screen is fixed at one end of a short brass tube and is looked at through a lens fixed at the other end of the tube. Viewed in this way, the surface of the screen is seen as a dark background, dotted with brilliant points of light which come and go with great rapidity. The number of points of light per unit area to be seen at one time falls off rapidly as the distance from the radium increases, and, at several centimetres 159distance, only an occasional one is seen. The experiment is extremely beautiful, and brings vividly before the observer the idea that the radium is shooting out a stream of projectiles, the impact of each of which on the screen is marked by a flash of light.
The scintillating points of light on the screen are the result of the impact of the α particles on its surface. If the radium is covered with a layer of foil of sufficient thickness to absorb all the α rays the scintillations cease. There is still a phosphorescence to be observed on the screen due to the β and γ rays, but this luminosity is not marked by scintillations to any appreciable extent. Sir William Crookes showed that the number of scintillations was about the same in vacuo as in air at atmospheric pressure. If the screen was kept at a constant temperature, but the radium cooled down to the temperature of liquid air, no appreciable difference in the number of scintillations was observed. If, however, the screen was gradually cooled to the temperature of liquid air, the scintillations diminished in number and finally ceased altogether. This is due to the fact that the screen loses to a large extent its power of phosphorescence at such a low temperature.
Not only are scintillations produced by radium, actinium, and polonium, but also by the emanations and other radio-active products which emit α rays. In addition, F. H. Glew[154] has found that they can be observed from the metal uranium, thorium compounds and various varieties of pitchblende. In order to show the scintillations produced by pitchblende, a flat surface was ground, and a transparent screen, whose lower surface was coated with zinc sulphide, placed upon it. Glew has designed a modified and very simple form of spinthariscope. A transparent screen, coated on one side with a thin layer of zinc sulphide, is placed in contact with the active material, and the scintillations observed by a lens in the usual way.
Since there is no absorption in the air, the luminosity is a maximum. The relative transparency of different substances placed between the active material and the screen may, in this way, be directly studied.
The production of scintillations appears to be a general property of the α rays from all radio-active substances. The 160scintillations are best shown with a zinc sulphide screen; but are also observed with willemite (zinc silicate), powdered diamond, and potassium platinocyanide (Glew, loc. cit.). If a screen of barium platinocyanide is exposed to the α rays from radium, the scintillations are difficult to observe, and the luminosity is far more persistent than for a zinc sulphide screen exposed under the same conditions. The duration of the phosphorescence in this case probably accounts for the absence of visible scintillations.
There can be no doubt that the scintillations result from the continuous bombardment of the sensitive screen by the α particles. Each of these particles moves with enormous velocity, and has a considerable energy of motion. On account of the ease with which these particles are stopped, most of this energy is given up at the surface of the screen, and a portion of the energy is in some way transformed into light. Zinc sulphide is very sensitive to mechanical shocks. Luminosity is observed if a penknife is drawn across the screen, or if a current of air is directed on to the screen. The disturbance effected by the impact of the α particle extends over a distance very large compared with the size of the impinging particle, so that the spots of light produced have an appreciable area. Recently Becquerel[155] has made an examination of the scintillations produced by different substances, and has concluded that the scintillations are due to irregular cleavages in the crystals composing the screen, produced by the action of the α rays. Scintillations can be mechanically produced by crushing a crystal. Tommasina[156] found that a zinc sulphide screen removed from the action of the radium rays for several days, showed the scintillations again when an electrified rod was brought near it.
The number of scintillations produced in zinc sulphide depends upon the presence of a slight amount of impurity and on its crystalline state. It can be shown that even with the most sensitive zinc sulphide screens, the number of scintillations is probably only a small fraction of the total number of α particles which fall upon it. It would appear that the crystals are in some way altered by the bombardment of the α particles, and that some of the crystals occasionally break up with emission of light[157].
161Although the scintillations from a particle of pure radium bromide are very numerous, they are not too numerous to be counted. Close to the radium, the luminosity is very bright, but by using a high power microscope the luminosity can still be shown to consist of scintillations. Since the number of scintillations probably bears no close relation to the number of α particles emitted, a determination of the number of scintillations would have no special physical significance. The relation between the number of α particles and the number of scintillations would probably be variable, depending greatly on the exact chemical composition of the sensitive substance and also upon its crystalline state.
97. Absorption of the α rays by matter. The α rays from the different radio-active substances can be distinguished from one another by the relative amounts of their absorption by gases or by thin screens of solid substances. When examined under the same conditions, the α rays from the active substances can be arranged in a definite order with reference to the amount of absorption in a given thickness of matter.
In order to test the amount of absorption of the α rays for different thicknesses of matter, an apparatus similar to that shown in Fig. 17, p. 98, was employed[158]. A thin layer of the active material was spread uniformly over an area of about 30 sq. cms., and the saturation current observed between two plates 3·5 cms. apart. With a thin layer[159] of active material, the ionization between the plates is due almost entirely to the α rays. The ionization due to the β and γ rays is generally less than 1% of the total.
The following table shows the variation of the saturation current between the plates due to the α rays from radium and polonium, with successive layers of aluminium foil interposed, each ·00034 cm. in thickness. In order to get rid of the ionization due to the β rays from radium, the radium chloride employed was dissolved in water and evaporated. This renders the active compound, for the time, nearly free from β rays.
162The initial current with 1 layer of aluminium over the active material is taken as 100. It will be observed that the current due
| Polonium. | Radium. | ||||
|---|---|---|---|---|---|
| Layers of aluminium | Current | Ratio of decrease for each layer | Layers of aluminium | Current | Ratio of decrease for each layer |
| 0 | 100 | 0 | 100 | ||
| ·41 | ·48 | ||||
| 1 | 41 | 1 | 48 | ||
| ·31 | ·48 | ||||
| 2 | 12·6 | 2 | 23 | ||
| ·17 | ·60 | ||||
| 3 | 2·1 | 3 | 13·6 | ||
| ·067 | ·47 | ||||
| 4 | ·14 | 4 | 6·4 | ||
| ·39 | |||||
| 5 | 0 | 5 | 2·5 | ||
| ·36 | |||||
| 6 | ·9 | ||||
| 7 | 0 |
to the radium rays decreases very nearly by half its value for each additional thickness until the current is reduced to about 6% of the maximum. It then decays more rapidly to zero. Thus, for radium, over a wide range, the current decreases approximately according to an exponential law with the thickness of the screen, or
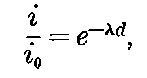
where i is the current for a thickness d, and i₀ the initial current. In the case of polonium, the decrease is far more rapid than would be indicated by the exponential law. By the first layer, the current is reduced to the ratio ·41. The addition of the third layer cuts the current down to a ratio of ·17. For most of the active bodies, the current diminishes slightly faster than the exponential law would lead one to expect, especially when the radiation is nearly all absorbed.
98. The increase of absorption of the α rays of polonium with the thickness of matter traversed has been very clearly shown in some experiments made by Mme Curie. The apparatus employed is shown in Fig. 34.
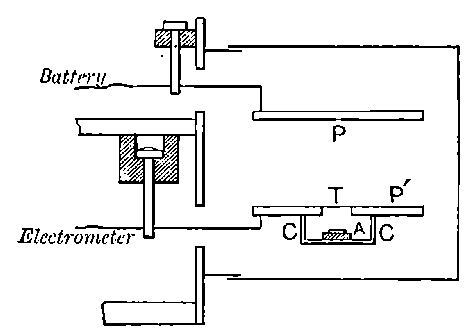
Fig. 34.
The saturation current was measured between two parallel plates PP´ 3 cms. apart. The polonium A was placed in the metal box CC, and the rays from it, after passing through an opening in the lower plate P´, covered with a layer of thin foil T, ionized the gas between the plates. For a certain distance AT, of 4 cms. or more, no appreciable current was observed between P and P´. As the distance AT was diminished, the current increased in a very sudden manner, so that for a small variation of the distance AT there was a large increase of current. With still further decrease of distance the current increases in a more regular manner. The results are shown in the following table, where the screen T consisted of one and two layers of aluminium foil respectively. The current due to the rays, without the aluminium screen, is in each case taken as 100.
| Distance AT in cms. | 3·5 | 2·5 | 1·9 | 1·45 | 0·5 |
|---|---|---|---|---|---|
| For 100 rays transmitted by one layer | 0 | 0 | 5 | 10 | 25 |
| For 100 rays transmitted by two layers | 0 | 0 | 0 | 0 | 0·7 |
The metallic screen thus cuts off a greater proportion of the rays the greater the distance of air which the radiations traverse. The effects are still more marked if the plates PP´ are close together. Results similar but not so marked are found if radium is substituted for the polonium.
It follows from these experiments that the ionization per unit volume, due to a large plate uniformly covered with the radio-active matter, falls off rapidly with the distance from the plate. At a distance of 10 cms. the α rays from uranium, thorium, or radium have been completely absorbed in the gas, and the small ionization then observed in the gas is due to the more penetrating β and γ rays. The relative amount of the ionization observed at 164a distance from the source will increase with the thickness of the layer of active matter, but will reach a maximum for a layer of a certain thickness. The greater proportion of the ionization, due to unscreened active matter, is thus entirely confined to a shell of air surrounding it not more than 10 cms. in depth.
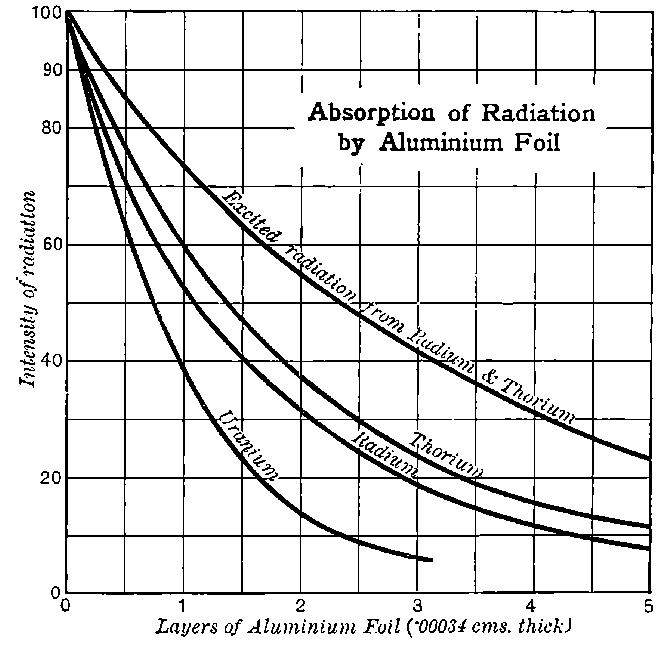
Fig. 35.
99. The α rays from different compounds of the same active element, although differing in amount, have about the same average penetrating power. Experiments on this point have been made by the writer[160] and by Owens[161]. Thus in comparing the relative power of penetration of the α rays from the different radio-elements, it is only necessary to determine the penetrating power for one compound of each of the radio-elements. Rutherford and Miss Brooks[162] have determined the amount of absorption of the α rays from the different active substances in their passage through successive layers of aluminium foil ·00034 cm. thick. The 165curves of absorption are given in Fig. 35. For the purpose of comparison in each case, the initial current with the bare active compound was taken as 100. A very thin layer of the active substance was used, and, in the case of thorium and radium, the emanations given off were removed by a slow current of air through the testing vessel. A potential difference of 300 volts was applied between the plates, which was sufficient to give the maximum current in each case.
Curves for the minerals organite and thorite were very nearly the same as for thoria.
For comparison, the absorption curves of the excited radiations of thorium and radium are given, as well as the curve for the radio-elements uranium, thorium, radium, and polonium. The α radiations may be arranged in the following order, as regards their power of penetration, beginning with the most penetrating.
The same order is observed for all the absorbing substances examined, viz., aluminium, Dutch metal, tinfoil, paper, and air and other gases. The differences in the absorption of the α rays from the active bodies are thus considerable, and must be ascribed either to a difference of mass or of velocity of the α particles or to a variation in both these quantities.
Since the α rays differ either in mass or velocity, it follows that they cannot be ascribed to any single radio-active impurity common to all radio-active bodies.
100. Absorption of the α rays by gases. The α rays from the different radio-active substances are quickly absorbed in their passage through a few centimetres of air at atmospheric pressure and temperature. In consequence of this, the ionization of the air, due to the α rays, is greatest near the surface of the radiating body and falls off very rapidly with the distance (see section 98).
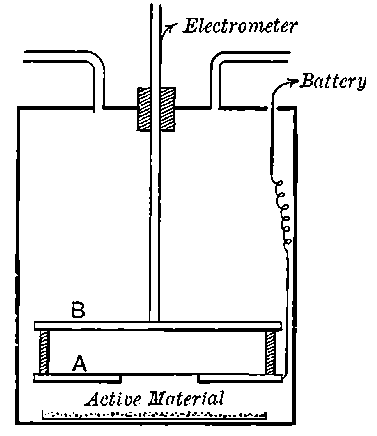
Fig. 36.
A simple method of determining the absorption in gases is shown in Fig. 36. The maximum current is measured between two parallel plates A and B kept at a fixed distance of 2 cms. apart, and then moved by means of a screw to different distances from the radio-active surface. The radiation from this active surface passed through a circular opening in the plate A, covered with thin aluminium foil, and was stopped by the upper plate. For observations on other gases besides air, and for examining the effect at different pressures, the apparatus is enclosed in an air-tight cylinder.
If the radius of the active surface is large compared with the distance of the plate A from it, the intensity of the radiation is approximately uniform over the opening in the plate A, and falls off with the distance x traversed according to an exponential law. Thus
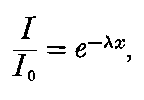
where λ is the “absorption constant” of the radiation for the gas under consideration[163]. Let
The energy of the radiation at the lower plate is then
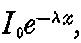
and at the upper plate
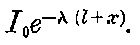
The total number of ions produced between the parallel plates A and B is therefore proportional to
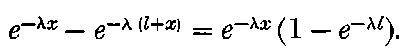
Since the factor
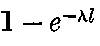
is a constant, the saturation current 167between A and B varies as
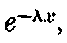
i.e. it decreases according to an exponential law with the distance traversed.
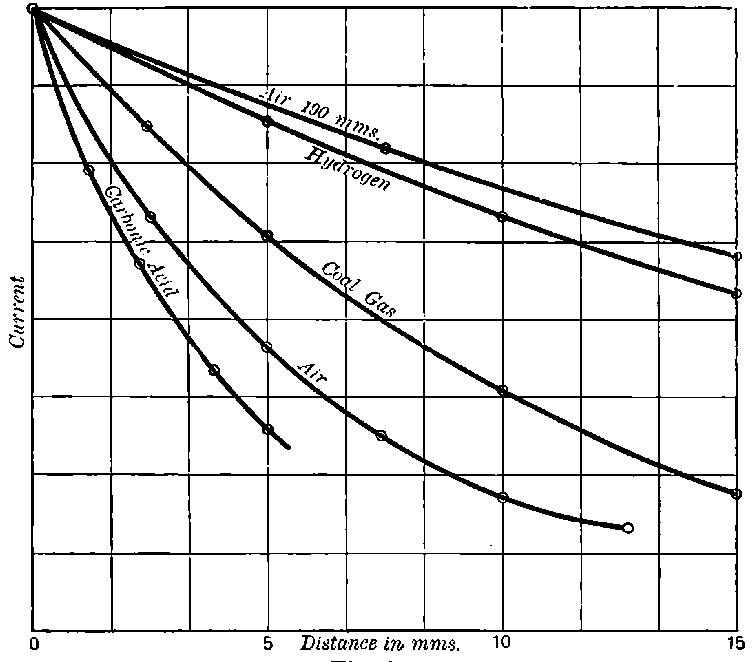
Fig. 37.
The variation of the current between A and B with the distance from a thin layer of uranium oxide is shown in Fig. 37 for different gases. The initial measurements were taken at a distance of about 3·5 mms. from the active surface. The actual values of this initial current were different for the different gases, but, for the purposes of comparison, the value is in each case taken as unity.
It will be seen that the current falls off with the distance approximately in a geometrical progression, a result which is in agreement with the simple theory given above. The distance through which the rays pass before they are absorbed is given below for different gases.
| Gas | Distance in mms. to absorb half of radiation |
|---|---|
| Carbonic acid | 3 |
| Air | 4·3 |
| Coal-gas | 7·5 |
| Hydrogen | 16 |
The results for hydrogen are only approximate, as the absorption is small over the distance examined.
168The absorption is least in hydrogen and greatest in carbonic acid, and follows the same order as the densities of the gases. In the case of air and carbonic acid, the absorption is proportional to the density, but this rule is widely departed from in the case of hydrogen. Results for the relative absorption by air of the α rays from the different active bodies are shown in Fig. 38.
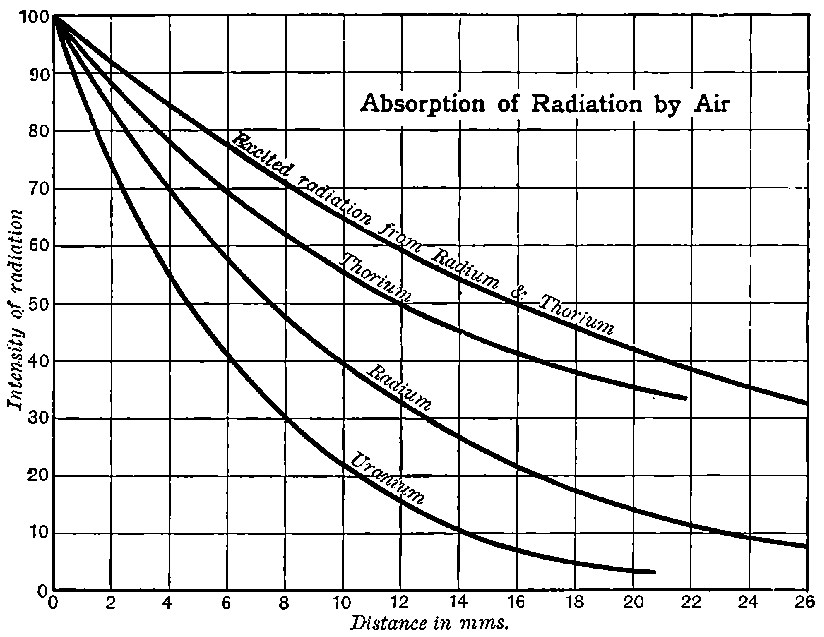
Fig. 38.
The initial observation was made about 2 mms. from the active surface, and the initial current is in each case taken as 100. The current, as in the case of uranium, falls off at first approximately in geometrical progression with the distance. The thickness of air, through which the radiation passes before the intensity is reduced to half value, is given below.
| Distance in mms. | |
|---|---|
| Uranium | 4·3 |
| Radium | 7·5 |
| Thorium | 10 |
| Excited radiation from Thorium and Radium | 16·5 |
The order of absorption by air of the radiations from the active substances is the same as the order of absorption by the metals and solid substances examined.
169101. Connection between absorption and density. Since in all cases the radiations first diminish approximately according to an exponential law with the distance traversed, the intensity I after passing through a thickness x is given by
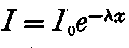
where λ is the absorption constant and I₀ the initial intensity.
The following table shows the value of λ with different radiations for air and aluminium.
| Radiation | λ for aluminium | λ for air |
|---|---|---|
| Excited radiation | 830 | ·42 |
| Thorium | 1250 | ·69 |
| Radium | 1600 | ·90 |
| Uranium | 2750 | 1·6 |
Taking the density of air at 20° C. and 760 mms. as 0·00120 compared with water as unity, the following table shows the value of λ divided by density for the different radiations.
| Radiation | Aluminium | Air |
|---|---|---|
| Excited radiation | 320 | 350 |
| Thorium | 480 | 550 |
| Radium | 620 | 740 |
| Uranium | 1060 | 1300 |
Comparing aluminium and air, the absorption is thus roughly proportional to the density for all the radiations. The divergence, however, between the absorption-density numbers is large when two metals like tin and aluminium are compared. The value of λ for tin is not much greater than for aluminium, although the density is nearly three times as great.
If the absorption is proportional to the density, the absorption in a gas should vary directly as the pressure, and this is found to be the case. Some results on this subject have been given by the writer (loc. cit.) for uranium rays between pressures of ¼ and 1 atmosphere. Owens (loc. cit.) examined the absorption of the α radiation in air from thoria between the pressures of 0·5 to 3 atmospheres and found that the absorption varied directly as the pressure.
The variation of absorption with density for the projected positive particles is thus very similar to the law for the projected negative particles and for cathode rays. The absorption, in both cases, depends mainly on the density, but is not in all cases directly 170proportional to it. Since the absorption of the α rays in gases is probably mainly due to the exhaustion of the energy of the rays by the production of ions in the gas, it seems probable that the absorption in metals is due to a similar cause.
102. Relation between ionization and absorption in gases. It has been shown (section 45) that if the α rays are completely absorbed in a gas, the total ionization produced is about the same for all the gases examined. Since the rays are unequally absorbed in different gases, there should be a direct connection between the relative ionization and the relative absorption. This is seen to be the case if the results of Strutt (section 45) are compared with the relative absorption constants (section 100).
| Gas | Relative absorption | Relative ionization |
|---|---|---|
| Air | 1 | 1 |
| Hydrogen | ·27 | ·226 |
| Carbon dioxide | 1·43 | 1·53 |
Considering the difficulty of obtaining accurate determinations of the absorption, the relative ionization in a gas is seen to be directly proportional to the relative absorption within the limits of experimental error. This result shows that the energy absorbed in producing an ion is about the same in air, hydrogen, and carbon dioxide.
103. Mechanism of the absorption of α rays by matter. The experiments, already described, show that the ionization of the gas, due to the α rays from a large plane surface of radio-active matter, falls off in most cases approximately according to an exponential law, until most of the rays are absorbed, whereupon the ionization decreases at a much faster rate. In the case of polonium, the ionization falls off more rapidly than is to be expected on the simple exponential law.
The ionization produced in the gas is due to the collision of the rapidly moving α particles with the molecules of the gas in their path. On account of its large mass, the α particle is a far more efficient ionizer than the β particle moving at the same speed. It can be deduced from the results of experiment that 171each projected α particle is able to produce about 100,000 ions in passing through a few centimetres of the gas before its velocity is reduced to the limiting value, below which it no longer ionizes the gas in its path.
Energy is required to ionize the gas, and this energy can only be obtained at the expense of the kinetic energy of the projected α particle. Thus it is to be expected that the α particle should gradually lose its velocity and energy of motion in its passage through the gas.
Since the rate of absorption of the α rays in gases is deduced from measurements of the ionization of the gas at different distances from the source of radiation, a knowledge of the law of variation of the ionizing power of the projected α particle with its speed is required in order to interpret the results. The experimental data on this question are, however, too incomplete to be applied directly to a solution of this question. Townsend[164] has shown that a moving electron produces ions in the gas after a certain limiting velocity is reached. The number of ions produced per centimetre of its path through the gas then rises to a maximum, and for still higher speeds continuously decreases. For example, Townsend found that the number of ions produced by an electron moving in an electric field was small at first for weak fields, but increased with the strength of the electric field to a maximum corresponding to the production of 20 ions per cm. of path in air at a pressure of 1 mm. of mercury. Durack[165] found that the electrons, generated in a vacuum tube, moving with a velocity of about 5 × 109 cms. per second produced a pair of ions every 5 cms. of path at 1 mm. pressure. In a later paper, Durack showed that for the electrons from radium, which are projected with a velocity greater than half the velocity of light, a pair of ions was produced every 10 cms. of path. The high speed electron from radium is thus a very inefficient ionizer and produces only about ¹⁄₁₀₀ of the ionization per unit path observed by Townsend for the slow moving electron.
104. In the case of the α particle, no direct measurements have been made upon the variation of the ionization with the 172velocity of the particle, so that the law of absorption of the rays cannot be deduced directly. An indirect attack upon the question has, however, been made recently by Bragg and Kleeman[166] who have formulated a simple theory to account for the experimental results which they have obtained upon the absorption of the α rays. The α particles from each simple type of radio-active matter are supposed to be projected with the same velocity, and to pass through a definite distance a in air at atmospheric pressure and temperature before they are all absorbed. As a first approximation the ionization per unit path is supposed to be the same over the whole length traversed before absorption, and to cease fairly suddenly at a definite distance from the source of radiation. This is in agreement with the observed fact that the ionization between parallel plates increases very rapidly when it approaches nearer than a certain distance to the radiant source. The range a depends upon the initial energy of motion of the α particle and will thus be different for different kinds of radio-active matter. If a thick layer of radio-active matter is employed, only the α particles from the surface have a range a. Those which reach the surface from a depth d have their range diminished by an amount ρd, where ρ is the density of the radio-active matter compared with air. This is merely an expression of the fact that the absorption of the α rays is proportional to the thickness and density of matter traversed. The rays from a thick layer of active matter will thus be complex, and will consist of particles of different velocity whose ranges have all values between 0 and a.
Suppose that a narrow pencil of α rays is emitted from a thick layer of radio-active material, and confined by metal stops as in Fig. 39.
Fig. 39.
The pencil of rays passes into an ionization vessel AB through a fine wire gauze A. The amount of ionization is to be determined between A and B for different distances h from the source of the rays R to the plate A.
173All the particles coming from a depth x of the material given by h = a – ρx will enter the ionization vessel. The number of ions produced in a depth dh of the ionization vessel is equal to nxdh, i.e. to
where n is a constant.
If the depth of the ionization vessel be b, the total number of ions produced in the vessel is
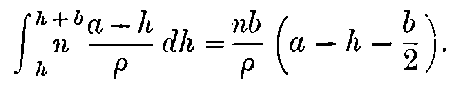
This supposes that the stream of particles passes completely across the vessel. If not, the expression becomes
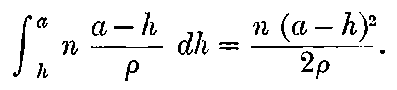
If the ionization in the vessel AB is measured, and a curve plotted showing its relation to h, the curve in the former case should be a straight line whose slope is nb/ρ and in the latter a parabola.
Thus if a thin layer of radio-active material is employed and a shallow ionization vessel, the ionization would be represented by a curve such as APM (Fig. 40), where the ordinates represent distances from the source of radiation, and the abscissae the ionization current between the plates AB.
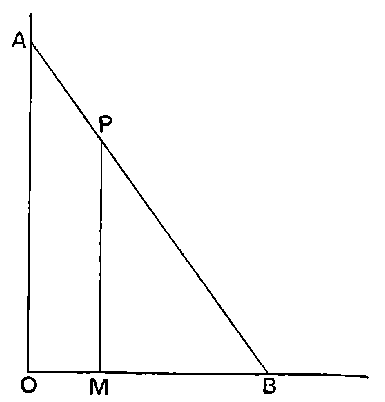
Fig. 40.
In this case, PM is the range of the α particles from the lowest layer of the radio-active matter. The current should be constant for all distances less than PM.
For a thick layer of radio-active matter, the curve should be a straight line such as APB.
Curves of the above character should only be obtained when definite cones of rays are employed, and where the ionization vessel is shallow and includes the whole cone of rays. In such a case the inverse square law need not be taken into account.
174In the experiments previously recorded (sections 99 and 100), the ionization was measured between parallel plates several centimetres apart for a large area of radio-active material. Such an arrangement was necessary at the time at which the experiments were made, as only weak radio-active material was available. Measurable electrical effects could not then be obtained with narrow cones of rays and shallow ionization vessels, but this disadvantage is removed by the advent of pure radium bromide as a source of radiation.
The interesting experiments described by Bragg and Kleeman show that the theoretical curves are approximately realized in practice. The chief difficulty experienced in the analysis of the experimental results was due to the fact that radium is a complex radio-active substance and contains four radio-active products each of which gives rise to α rays which have different ranges. The general character of the results obtained from radium are shown graphically in Fig. 41, curves A, B, C, D.
Fig. 41.
The ordinates represent the distance between the radium and the gauze of the testing vessel; the abscissae the current in the ionization vessel in arbitrary units. Five milligrams of radium bromide were used, and the depth of the ionization vessel was 175about 5 mms. Curve A is for a cone of rays of angle 20°. The initial current at a distance of 7 cms. is due to the β and γ rays and natural leak. This curve is initially parabolic, and then is made up of two straight lines. Curve B is for a smaller cone, and shows the straight line character of the curve to within a short distance of the radium. Curve C was obtained under the same condition as curve A, but with a layer of gold beater’s skin placed over the radium. The effect of this is to reduce all the ordinates of curve A by the same quantity. This is to be expected on the simple theory already considered. Curve D was obtained when the radium was heated so as to get rid of the emanation and its products. The α particles of greatest range are quite absent and the curve is simpler in character.
Fig. 42.
The complex character of the radium curves are more clearly brought out by a careful examination of a portion of the curve at distances between 2 and 5 cms. from the radium, using an ionization vessel of depth only 2 mms. The results are shown in Fig. 42, where the curve is seen to consist approximately of 176four straight lines of different slopes represented by PQ, QR, RS, ST.
Such a result is to be expected, for it will be shown later that four distinct α ray products exist in radium when in radio-active equilibrium. Each of these products of radium emits an equal number of α particles per second, but the range of each is different. If a1 is the range of one stream, a2 of another, the ionization in the vessel AB, when two streams enter the vessel, should be
i.e.
Thus the slope of the curve should in this case be 2nb/ρ, while if only one stream enters, it should be nb/ρ. When three reach it, the slope should be 3nb/ρ and for four 4nb/ρ. These results are realized fairly closely in practice. The curve (Fig. 42) consists of four parts, whose slopes are in the proportion 16, 34, 45, 65, i.e. very nearly in the ratio 1, 2, 3, 4.
Experiments were also made with very thin layers of radium bromide, when, as we have seen (Fig. 40) a very different shape of curve is to be expected. An example of the results is shown in Fig. 43, curves I., II. and III. Curve I. is obtained from radium bromide which has been heated to drive off the emanation, and curves II. and III. from the same substance several days later, when the emanation was again accumulating. The portion PQ, which is absent in the first curve, is probably due to the “excited” activity produced by the emanation. By careful examination of the successive changes in the curves after the radium has been heated to drive off the emanation, it is possible to tell the range of the α rays from each of the different products, and this has been done to some extent by Bragg and Kleeman.
It will be seen later that the results here obtained support in a novel way the theory of radio-active changes which has been advanced from data of quite a different character.
The inward slope of the curve in Fig. 43 due to the radium indicates that the α particles become more efficient ionizers as 177their velocity decreases. This is in agreement with observations on the β rays. In some cases Bragg also observed that the α particles are the most efficient ionizers just before they lose their power of ionizing the gas.
Fig. 43.
Thus we may conclude from these experiments that the α particles from a simple radio-active substance traverse a definite distance in air, at a definite pressure and temperature, and that the ionization ends fairly abruptly. If the rays traverse a sheet of metal, the effective range of ionization is diminished by a distance corresponding to ρd, where ρ is the density of the material compared with air and d its thickness. The α rays from a thick layer of a simple radio-active substance consist of α particles of 178different velocities, which have ranges in air lying between 0 and the maximum range. The ionization of the particles per unit path is greatest near the end of its range, and decreases somewhat as we approach the radiant source. A complex source of rays like radium gives out four types of rays, each of which has a different but distinct range.
From this theory it is possible to calculate approximately the decrease of current to be observed when sheets of metal foil are placed over a large area of radio-active substance. This is the method that has been employed to obtain the curves of Figs. 35 and 38.
Suppose a very thin layer of simple radio-active matter is employed (for example a bismuth plate covered with radio-tellurium or a metal plate made active by exposure to the presence of the thorium or radium emanations) and that the ionization vessel is of sufficient depth to absorb the α rays completely.
Let d be the thickness of the metal plate, ρ its density compared with air. Consider a point P close to the upper side of the plate. The range of the particles moving from a point, when the path makes an angle θ with the normal at P, is a – ρd sec θ, where a is the range in air. The rays coming from points such that the paths make an angle with the normal greater than
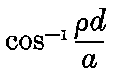
will thus be absorbed in the plate. By integrating over the circular area under the point P, it is easy to show that the total ionization in the vessel is proportional to
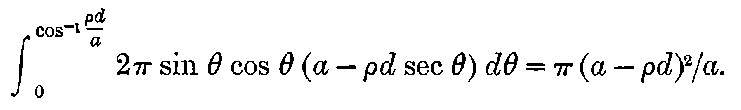
The curves showing the relation between current and distance of metal traversed should thus be parabolic with respect to d. This is approximately the case for a simple substance like radio-tellurium. The curve for a thick layer of radium would be difficult to calculate on account of the complexity of the rays, but we know from experiment that it is approximately exponential. An account of some recent investigations made to determine the range of velocity over which the α particle is able to ionize the gas is given in Appendix A. The results there given strongly support the theory of absorption of the α rays discussed above.
105. In addition to the α and β rays, the three active substances, uranium, thorium, and radium, all give out a radiation of an extraordinarily penetrating character. These γ rays are considerably more penetrating than the X rays produced in a “hard” vacuum tube. Their presence can readily be observed for an active substance like radium, but is difficult to detect for uranium and thorium unless a large quantity of active material is used. Villard[167], using the photographic method, first drew attention to the fact that radium gave out these very penetrating rays, and found that they were non-deviable by a magnetic field. This result was confirmed by Becquerel[168].
Using a few milligrams of radium bromide, the γ rays can be detected in a dark room by the luminosity they excite in the mineral willemite or a screen of platinocyanide of barium. The α and β rays are completely absorbed by placing a thickness of 1 centimetre of lead over the radium, and the rays which then pass through the lead consist entirely of γ rays. The very great penetrating power of these rays is easily observed by noting the slight diminution of the luminosity of the screen when plates of metal several centimetres thick are placed between the radium and the screen. These rays also produce ionization in gases and are best investigated by the electrical method. The presence of the γ rays from 30 mgrs. of radium bromide can be observed in an electroscope after passing through 30 cms. of iron.
106. Absorption of the γ rays. In an examination of the active substances by the electrical method, the writer[169] found that both uranium and thorium gave out γ rays in amount roughly proportional to their activity. An electroscope of the type shown in Fig. 12 was employed. This was placed on a large lead plate ·65 cm. thick, the active substance being placed in a closed vessel beneath.
180The discharge due to the natural ionization of the air in the electroscope was first observed. The additional ionization due to the active substance must be that produced by rays which have passed through the lead plate and the walls of the electroscope. The following table shows that the discharge due to these rays decreases approximately according to an exponential law with the thickness of lead traversed.
| Thickness of lead | Rate of discharge |
|---|---|
| ·62 cms. | 100 |
| „ + ·64 cms. | 67 |
| „ + 2·86 „ | 23 |
| „ + 5·08 „ | 8 |
Using 100 grs. of uranium and thorium, the discharge due to the rays through 1 cm. of lead was quite appreciable, and readily measured. The results showed that the amount of γ rays was about the same for equal weights of thorium and uranium oxides. The penetrating power was also about the same as for the radium rays.
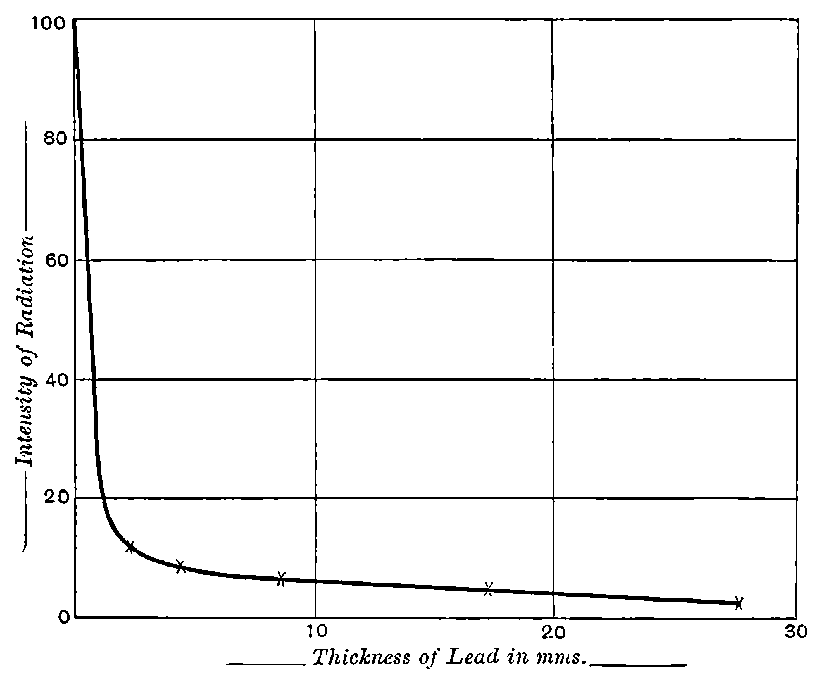
Fig. 44.
181The writer showed that the absorption of the γ rays from radium was approximately proportional to the density of the substance traversed. A more detailed examination of the absorption of these rays in various substances has been recently made by McClelland[170]. The curve (Fig. 44) shows the decrease of the ionization current in a testing vessel due to the β and γ rays with successive layers of lead. It is seen that the β rays are almost completely stopped by 4 mms. of lead; the ionization is then due entirely to the γ rays.
In order to leave no doubt that all the β rays were absorbed, the radium was covered with a thickness of 8 mms. of lead, and measurements of the coefficient of absorption λ were made for additional thicknesses. The average value of λ was calculated from the usual equation
where d is the thickness of matter traversed. The following table shows the value of λ, (I) for the first 2·5 mms. of matter traversed (after initially passing through 8 mms. of lead), (II) for the thickness 2·5 to 5 mms., (III) for 5 to 10 mms., (IV) 10 to 15 mms.
TABLE A.
| Substance | I | II | III | IV |
|---|---|---|---|---|
| Platinum | 1·167 | |||
| Mercury | ·726 | ·661 | ·538 | ·493 |
| Lead | ·641 | ·563 | ·480 | ·440 |
| Zinc | ·282 | ·266 | ·248 | ·266 |
| Aluminium | ·104 | ·104 | ·104 | ·104 |
| Glass | ·087 | ·087 | ·087 | ·087 |
| Water | ·034 | ·034 | ·034 | ·034 |
In the above table, the absorption in aluminium, glass and water was too small to determine with accuracy the variation of λ with distance traversed. It will be observed that, for the denser substances, the coefficient of absorption decreases with the distance through which the rays have passed. This indicates that the rays are heterogeneous. The variation of λ is more marked in heavy substances.
182Table B gives the values of λ divided by density for the above numbers. If the absorption were directly proportional to the density, the quotient would be the same in all cases.
TABLE B.
λ divided by density.
| Substance | I | II | III | IV |
|---|---|---|---|---|
| Platinum | ·054 | |||
| Mercury | ·053 | ·048 | ·039 | ·036 |
| Lead | ·056 | ·049 | ·042 | ·037 |
| Zinc | ·039 | ·037 | ·034 | ·033 |
| Aluminium | ·038 | ·038 | ·038 | ·038 |
| Glass | ·034 | ·034 | ·034 | ·034 |
| Water | ·034 | ·034 | ·034 | ·034 |
The numbers in column I vary considerably, but the agreement becomes closer in the succeeding columns, until in column IV the absorption is very nearly proportional to the density.
It is seen that the absorption of all three types of rays from radio-active substances is approximately proportional to the density of the substance traversed—a relation first observed by Lenard for the cathode rays. This law of absorption thus holds for both positively and negatively electrified particles projected from the radio-active substances, and also for the electromagnetic pulses which are believed to constitute the γ rays; although the absorption of the α rays, for example, is 10,000 times greater than for the γ rays. We have seen in section 84 that the value of the absorption constant λ for lead is 122 for the β rays from uranium. The value for the γ rays from radium varies between ·64 and ·44, showing that the γ rays are more than 200 times as penetrating as the β rays.
107. Nature of the rays. In addition to their great penetrating power, the γ rays differ from the α and β rays in not being deflected to an appreciable degree by a magnetic or electric field. In a strong magnetic field, it can be shown, using the photographic method, that there is an abrupt discontinuity between the β and γ rays, for the former are bent completely away 183from the latter. This indicates that, as regards the action of a magnetic field, there is no gradual transition of magnetic properties between the β and γ rays. Paschen[171] has examined the γ rays in a very intense magnetic field, and, from the absence of deflection of these rays, has calculated that, if they consist of electrified particles carrying an ionic charge, and projected with a velocity approaching that of light, their apparent mass must be at least 45 times greater than that of the hydrogen atom.
It now remains for us to consider whether the γ rays are corpuscular in character, or whether they are a type of electromagnetic pulse in the ether similar to Röntgen rays. They resemble Röntgen rays in their great penetrating power and in their absence of deflection in a magnetic field. Earlier experiments seemed to indicate an important difference between the action of γ and X rays. It is well known that ordinary X rays produce much greater ionization in gases such as sulphuretted hydrogen and hydrochloric acid gas, than in air, although the differences in density are not large. For example, exposed to X rays, sulphuretted hydrogen has six times the conductivity of air, while with γ rays the conductivity only slightly exceeds that of air. The results obtained by Strutt, in this connection, have already been given in section 45. It is there shown that the relative conductivity of gases exposed to γ rays (and also to α and β rays) is, in most cases, nearly proportional to their relative densities; but, under X rays, the relative conductivity for some gases and vapours is very much greater than for the γ rays. It must be remembered, however, that the results obtained by Strutt were for “soft X rays,” whose penetrating power was very much less than that of the γ rays. In order to see if the relative conductivity of gases produced by X rays depended upon their penetrating power, A. S. Eve[172] made some experiments with a very “hard” X ray bulb, which gave an unusually penetrating type of rays.
The results of the measurements are shown in the table below, where the conductivity for each type of rays is expressed relative to air as unity. The results obtained for “soft” X rays by Strutt and by Eve for γ rays are added for comparison.
184It is seen that the hard rays show a much closer agreement than the soft rays with the density law found for the γ rays. The high values previously obtained for the vapours of chloroform and carbon tetrachloride are greatly reduced, and are very nearly the same as for the γ rays. On the other hand, the vapour of methyl iodide is an exception, and still shows a high conductivity. The γ rays were, however, forty times as penetrating as the hard X rays, and it is probable that the value of methyl iodide would be reduced with still more penetrating X rays.
Relative conductivities of gases.
| Gas | Relative Density | “Soft” X rays | “Hard” X rays | γ rays |
|---|---|---|---|---|
| Hydrogen | ·07 | ·11 | ·42 | ·19 |
| Air | 1·0 | 1·0 | 1·0 | 1·0 |
| Sulphuretted Hydrogen | 1·2 | 6 | ·9 | 1·23 |
| Chloroform | 4·3 | 32 | 4·6 | 4·8 |
| Methyl Iodide | 5·0 | 72 | 13·5 | 5·6 |
| Carbon Tetrachloride | 5·3 | 45 | 4·9 | 5·2 |
The hard X rays were found to give far more secondary radiation than the γ rays, but this effect is probably also a function of the penetrating power of the primary rays. It will be seen later (section 112) that γ rays give rise to a secondary radiation of the β ray type. This has also been observed for the X rays.
Considering the experimental evidence as a whole, there is undoubtedly a very marked similarity between the properties of γ and X rays. The view that the γ rays are a type of very penetrating X rays, also receives support from theoretical considerations. We have seen (section 52) that the X rays are believed to be electromagnetic pulses, akin in some respects to short light waves, which are set up by the sudden stoppage of the cathode ray particles. Conversely, it is also to be expected that X rays will be produced at the sudden starting, as well as at the sudden stopping, of electrons. Since most of the β particles from radium are ejected from the radium atom with velocities much greater than the cathode particles in a vacuum tube, X rays of a very penetrating character will arise. But 185the strongest argument in support of this view is derived from an examination of the origin and connection of the β and γ rays from radio-active substances. It will be shown later that the α ray activity observed in radium arises from several disintegration products, stored up in the radium, while the β and γ rays arise only from one of these products named radium C. It is found, too, that the activity measured by the γ rays is always proportional to the activity measured by the β rays, although by separation of the products the activity of the latter may be made to undergo great variations in value.
Thus the intensity of the γ rays is always proportional to the rate of expulsion of β particles, and this result indicates that there is a close connection between the β and γ rays. Such a result is to be expected if the β particle is the parent of the γ ray, for the expulsion of each electron from radium will give rise to a narrow spherical pulse travelling from the point of disturbance with the velocity of light.
108. There is another possible hypothesis in regard to the nature of these rays. It has been shown (sections 48 and 82) that the apparent mass of an electron increases as the speed of light is approached; theoretically it should be very great when the velocity of the electron is exceedingly close to the velocity of light. In such a case, a moving electron would be difficult to deflect by a magnetic or electric field.
The view that the γ rays are electrons carrying a negative charge and moving with a velocity nearly equal to that of light has recently been advocated by Paschen[173]. He concluded from experiment that the γ rays like the β rays carried a negative charge. We have seen (section 85) that Seitz also observed that a small negative charge was communicated to bodies on which the γ rays impinged, but the magnitude of this charge was much smaller than that observed by Paschen. I do not think that much weight can be attached to observations that a small positive or negative charge is communicated to bodies on which the γ rays fall, for it will be shown later that a strong secondary radiation, 186consisting in part of electrons, is set up during the passage of the γ rays through matter. It is not improbable that the small charge observed is not a direct result of the charge carried by the γ rays, but is an indirect effect due to the secondary radiations emitted from the surface of bodies. There is no doubt that a thick lead vessel, completely enclosing a quantity of radium, acquires a small positive charge, but this result would follow whether the γ rays carry a charge or not, since the secondary radiations from the lead surface consist of projected particles which carry with them a negative charge.
On this corpuscular theory of the nature of the γ rays, each electron must have a large apparent mass, or otherwise it would be appreciably deflected by an intense magnetic field. The energy of motion of the electron must, in consequence, be very great, and, if the number of the electrons constituting the γ rays is of the same order of magnitude as the number of the β particles, a large heating effect is to be expected when the γ rays are stopped in matter. Paschen[174] made some experiments on the heat emission of radium due to the γ rays; he concluded that the γ rays were responsible for more than half of the total heat emission of radium and carried away energy at the rate of over 100 gram calories per hour per gram of radium. This result was not confirmed by later experiments of Rutherford and Barnes[175], who found that the heating effect of the γ rays could not be more than a few per cent. of the total heat emission of radium. These results will be considered later in chapter XII.
The weight of evidence, both experimental and theoretical, at present supports the view that the γ rays are of the same nature as the X rays but of a more penetrating type. The theory that the X rays consist of non-periodic pulses in the ether, set up when the motion of electrons is arrested, has found most favour, although it is difficult to provide experimental tests to decide definitely the question. The strongest evidence in support of the wave nature of the X rays is derived from the experiments of Barkla[176], who found that the amount of secondary radiation set up by the X rays 187on striking a metallic surface depended on the orientation of the X ray bulb. The rays thus showed evidence of a one-sidedness or polarization which is only to be expected if the rays consist of a wave motion in the ether.
109. Production of secondary rays. It has long been known that Röntgen rays, when they impinge on solid obstacles, produce secondary rays of much less penetrating power than the incident rays. This was first shown by Perrin and has been investigated in detail by Sagnac, Langevin, Townsend and others. Thus it is not surprising that similar phenomena should be observed for the radiation from radio-active substances. By means of the photographic method, Becquerel[177] has made a close study of the secondary radiations produced by radio-active substances. In his earliest observations, he noticed that radiographs of metallic objects were always surrounded by a diffuse border. This effect is due to the secondary rays set up by the incident rays at the surface of the screen.
The secondary rays produced by the α rays are very feeble. They are best shown by polonium, which gives out only α rays, so that the results are not complicated by the action of the β rays. Strong secondary rays are set up at the point of impact of the β or cathodic rays. Becquerel found that the magnitude of this action depended greatly on the velocity of the rays. The rays of lowest velocity gave the most intense secondary action, while the penetrating rays gave, in comparison, scarcely any secondary effect. In consequence of the presence of this secondary radiation, the photographic impression of a screen pierced with holes is not clear and distinct. In each case there is a double photographic impression, due to the primary rays and the secondary rays set up by them.
These secondary rays are deviable by a magnetic field, and in turn produce tertiary rays and so on. The secondary rays are in all cases more readily deviated and absorbed than the primary rays, 188from which they arise. The very penetrating γ rays give rise to secondary rays, which cause intense action on the photographic plate. When some radium was placed in a cavity inside a deep lead block, rectangular in shape, besides the impression due to the direct rays through the lead, Becquerel observed that there was also a strong impression due to the secondary rays emitted from the surface of the lead. The action of these secondary rays on the plate is so strong that the effect on the plate is, in many cases, increased by adding a metal screen between the active material and the plate.
The comparative photographic action of the primary and secondary rays cannot be taken as a relative measure of the intensity of their radiations. For example, only a small portion of the energy of the β rays is in general absorbed in the sensitive film. Since the secondary rays are far more easily absorbed than the primary rays, a far greater proportion of their energy is expended in producing photographic action than in the case of the primary rays. It is thus not surprising that the secondary rays set up by the β and γ rays may in some cases produce a photographic impression comparable with, if not greater than, the effect of the incident rays.
On account of these secondary rays, radiographs produced by the β rays of radium in general show a diffuse border round the shadow of the object. For this reason radiographs of this kind lack the sharpness of outline of X ray photographs.
110. Secondary radiation produced by α rays. Mme Curie[178] has shown by the electric method that the α rays of polonium produce secondary rays. The method adopted was to compare the ionization current between two parallel plates, when two screens of different material, placed over the polonium, were interchanged.
These results show that the α rays of polonium are modified in passing through matter, and that the amount of secondary rays set up varies with screens of different material. Mme Curie, using the same method, was unable to observe any such effect for the β rays of radium. The production of secondary rays by the β rays of 189radium is, however, readily shown by the photographic method. We have already seen (section 93) that very low velocity electrons accompany the α rays from radium or radio-tellurium spread on a metal plate. These electrons are probably liberated when the α rays escape from or impinge upon matter, and the number emitted depends upon the kind of matter used as a screen. The differences shown in the above table when the screens were interchanged are explained simply in this way.
| Screens employed | Thickness in mms. | Current observed |
|---|---|---|
| Aluminium | 0·01 | |
| Cardboard | 0·005 | 17·9 |
| Cardboard | 0·005 | |
| Aluminium | 0·01 | 6·7 |
| Aluminium | 0·01 | |
| Tin | 0·005 | 150 |
| Tin | 0·005 | |
| Aluminium | 0·01 | 126 |
| Tin | 0·005 | |
| Cardboard | 0·005 | 13·9 |
| Cardboard | 0·005 | |
| Tin | 0·005 | 4·4 |
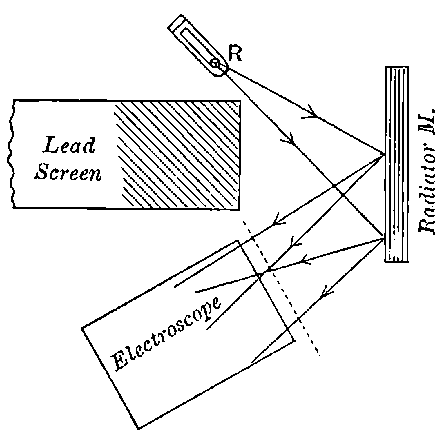
Fig. 45.
111. Secondary rays produced by β and γ rays. An examination of the amount and character of the secondary radiation emitted by various substances, when exposed to the 190β and γ rays of radium, has recently been made by A. S. Eve[179]. The general experimental method employed is shown in Fig. 45.
The electroscope (Fig. 45) was placed behind a lead screen 4·5 cms. thick, which stopped all the β rays and absorbed the greater proportion of the γ rays from the radium tube placed at R. On bringing near a plate of matter M, the primary rays fell upon it and some of the secondary rays, emitted in all directions, passed into the side of the electroscope, which was covered with aluminium foil of thickness ·05 mm. Before the plate M was placed in position the rate of discharge of the electroscope was due to the natural leak and the γ rays from R, and the secondary radiation from the air. On bringing the radiator M into position, the rate of discharge was much increased, and the difference between the rate of movement of the gold-leaf in the two cases was taken as a measure of the amount of secondary rays from M. The absorption of the secondary rays was tested by placing an aluminium plate ·85 mm. thick before the face of the electroscope.
The secondary rays were found to be fairly homogeneous, for the intensity fell off according to an exponential law with the distance traversed. The value of the absorption constant λ was determined from the usual equation
where d is the thickness of the screen. The table given below shows the results obtained when thick plates of different substances of the same dimensions were placed in a definite position at M. The secondary radiation from fluids was obtained by a slight alteration of the experimental arrangements.
Thirty milligrammes of radium bromide were used, and the results are expressed in terms of the number of scale divisions passed over per second by the gold-leaf.
It will be noticed that the amount of secondary radiation follows in most cases the same order as the densities, and is greatest for mercury. The value of (secondary radiation)/density is not a constant, but varies considerably, being greatest for light substances. The absorption constant of the secondary rays from different radiators is not very different, with the exception 191of substances such as granite, brick, and cement, which give out secondary rays of nearly twice the penetrating power of other substances.
β and γ rays.
| Radiator | Density | Secondary Radiation | Sec. Rad. / Density | Aluminium ·085 cm. λ |
|---|---|---|---|---|
| Mercury | 13·6 | 147 | 10·8 | |
| Lead | 11·4 | 141 | 12·4 | 18·5 |
| Copper | 8·8 | 79 | 9·0 | 20 |
| Brass | 8·4 | 81 | 9·6 | 21 |
| Iron (wrought) | 7·8 | 75 | 9·6 | 20 |
| Tin | 7·4 | 73 | 9·9 | 20·3 |
| Zinc | 7·0 | 79 | 11·3 | |
| Granite | 2·7 | 54 | 20·0 | 12·4 |
| Slate | 2·6 | 53 | 20·4 | 12·1 |
| Aluminium | 2·6 | 42 | 16·1 | 24 |
| Glass | 2·5 | 44 | 17·6 | 24 |
| Cement | 2·4 | 47 | 19·6 | 13·5 |
| Brick | 2·2 | 49 | 22·3 | 13·0 |
| Ebonite | 1·1 | 32 | 29·1 | 26 |
| Water | 1·0 | 24 | 24·0 | 21 |
| Ice | ·92 | 26 | 28·2 | |
| Paraffin solid | ·9 | 17 | 18·8 | 21 |
| „ liquid | ·85 | 16 | 18·8 | |
| Mahogany | ·56 | 21·4 | 38·2 | 23 |
| Paper | ·4? | 21·0 | 52 | 22 |
| Millboard | ·4? | 19·4 | 48 | 20·5 |
| Papier-mâché | ... | 21·9 | ||
| Basswood | ·36 | 20·7 | 57 | 22 |
| Pine | ·35 | 21·8 | 62 | 21 |
| X ray screen | 75·2 | 23·6 |
The secondary radiation not only comes from the surface of the radiator but from a considerable depth. The amount of secondary rays increases with the thickness of the radiator, and, in the case of glass and aluminium, reaches a practical maximum for a plate about 3 mms. thick.
In the above table, the secondary radiation arises from both the β rays and γ rays together. When the β rays were cut off by a layer of lead 6·3 mms. thick, placed between the radium and the radiator, the effect on the electroscope was reduced to less than 20 per cent. of its former value, showing that the β rays supplied 192more than 80 per cent. of the secondary radiation. The following table shows the relative amount of secondary rays from different substances when exposed to β and γ rays together and to γ rays alone. The amount from lead in each case is taken as a standard and equal to 100. The amount of secondary radiation found by Townsend from soft X rays is added for comparison.
Secondary Radiations.
| Radiator | β and γ rays | γ rays | Röntgen |
|---|---|---|---|
| Lead | 100 | 100 | 100 |
| Copper | 57 | 61 | 291 |
| Brass | 58 | 59 | 263 |
| Zinc | 57 | ... | 282 |
| Aluminium | 30 | 30 | 25 |
| Glass | 31 | 35 | 31 |
| Paraffin | 12 | 20 | 125 |
It will be observed that the relative amounts are about the same for the γ rays alone as for the β and γ rays together. On the other hand, the amount of secondary radiation set up by X rays is very different, lead for example giving much less than brass or copper. The secondary rays from the γ rays alone are slightly less penetrating than for the β and γ rays together, but are far more penetrating than the secondary radiation from the X rays examined by Townsend.
The amount of secondary radiation set up by the β and γ rays is mainly independent of the state of the surface of the radiator. About the same amount is obtained from iron as from iron filings; from liquid as from solid paraffin; and from ice as from water[180].
Becquerel has shown that the secondary rays set up by the β rays are deflected by a magnet and consist of negatively 193charged particles (electrons). It has been pointed out in section 52 that the cathode rays are diffusely reflected from the metal on which they fall. These secondary rays consist in part of electrons moving with about the same velocity as the primary, and in part of some electrons with a much slower speed. The secondary rays set up by the β rays of radium have on an average less penetrating power than the primary rays, and consequently less velocity than the primary rays. It must be remembered that the β rays from radium are very complex, and consist of electrons projected with a considerable range of velocities. The secondary rays are, on an average, certainly more penetrating than the most easily absorbed β rays emitted from radium, and probably move with a velocity of about half that of light.
It is still uncertain whether the secondary rays are produced by the action of the primary rays on matter, or whether they consist of a portion of the primary rays whose direction of motion has been deflected in their passage through matter, so that they emerge again with diminished velocity from the surface.
112. Magnetic deflection of secondary rays from γ rays. It has been seen that the secondary rays set up by the γ rays alone are very similar in character to those caused by the β rays. This result was still further confirmed by Eve, who showed that the secondary rays produced by the γ rays are readily deflected by a magnetic field. The experimental arrangement is shown in Fig. 46.
Fig. 46.
A small electroscope was mounted on one side of a lead platform 1·2 cms. thick, which rested on a lead cylinder 10 cms. high and 10 cms. in diameter. The radium was placed at the bottom of a hole reaching to the centre of the cylinder.
On applying a strong magnetic field, at right angles to the plane of the paper, so as to bend the secondary rays from the platform towards the electroscope, the rate of discharge was much increased. On reversing the field, the effect was much diminished. Since the γ rays are not themselves deflected by a magnetic field, this result shows that the secondary radiation is quite different in character from the primary rays, and consists of electrons projected with a velocity (deduced from the penetrating power) of about half 194the velocity of light. We have already pointed out that the emission of electrons from a substance traversed by the rays will account sufficiently well for the charge observed by Paschen, without the necessity of assuming that the γ rays carry a negative charge of electricity.
The secondary radiation set up by Röntgen rays, like that due to the β and γ rays, consists in part of electrons projected with considerable velocity. These three types of rays seem about equally efficient in causing the expulsion of electrons from the substance through which they pass. We have seen that the X and γ rays are, in all probability, electromagnetic pulses set up by the sudden starting or stopping of electrons, and, since these rays in turn cause the removal of electrons, the process appears to be reversible. Since the β rays pass through some thickness of matter before their energy of motion is arrested, theory would lead us to expect that a type of soft X rays should be generated in the absorbing matter.
113. Comparison of the ionization produced by the α and β rays. With unscreened active material the ionization produced between two parallel plates, placed as in Fig. 17, is mainly due to the α rays. On account of the slight penetrating power of the α rays, the current due to them practically reaches a maximum with a small thickness of radio-active material. The following saturation currents were observed[181] for different thicknesses of uranium oxide between parallel plates sufficiently far apart for all the α rays to be absorbed in the gas between them.
Surface of uranium oxide 38 sq. cms.
| Weight of uranium oxide in grammes per sq. cm. of surface | Saturation current in amperes per sq. cm. of surface |
|---|---|
| . | |
| ·0036 | 1·7 × 10-13 |
| ·0096 | 3·2 × 10-13 |
| ·0189 | 4·0 × 10-13 |
| ·0350 | 4·4 × 10-13 |
| ·0955 | 4·7 × 10-13 |
The current reached about half its maximum value for a weight of oxide ·0055 gr. per sq. cm. If the α rays are cut off by a metallic screen, the ionization is then mainly due to the β rays, since the ionization produced by the γ rays is small in comparison. For the β rays from uranium oxide it has been shown (section 86) that the current reaches half its maximum value for a thickness of 0·11 gr. per sq. cm.
Meyer and Schweidler[182] have found that the radiation from a water solution of uranium nitrate is very nearly proportional to the amount of uranium present in the solution.
On account of the difference in the penetrating power of the α and β rays, the ratio of the ionization currents produced by them 196depends on the thickness of the radio-active layer under examination. The following comparative values of the current due to the α and β rays were obtained for very thin layers of active matter[183]. A weight of ⅒ gramme of fine powder, consisting of uranium oxide, thorium oxide, or radium chloride of activity 2000, was spread as uniformly as possible over an area of 80 sq. cms. The saturation current was observed between parallel plates 5·7 cms. apart. This distance was sufficient to absorb most of the α rays from the active substances. A layer of aluminium ·009 cm. thick absorbed all the α rays.
| Current due to α rays | Current due to β rays | Ratio of currents β/α | |
|---|---|---|---|
| Uranium | 1 | 1 | ·0074 |
| Thorium | 1 | ·27 | ·0020 |
| Radium | 2000 | 1350 | ·0033 |
In the above table the saturation current due to the α and β rays of uranium is, in each case, taken as unity. The third column gives the ratio of the currents observed for equal weights of substance. The results are only approximate in character, for the ionization due to a given weight of substance depends on its fineness of division. In all cases, the current due to the β rays is small compared with that due to the α rays, being greatest for uranium and least for thorium. As the thickness of layer increases, the ratio of currents β/α steadily increases to a constant value.
114. Comparison of the energy radiated by the α and β rays. It has not yet been found possible to measure directly the energy of the α and β rays. A comparison of the energy radiated in the two forms of rays can, however, be made indirectly by two distinct methods.
If it be assumed that the same amount of energy is required to produce an ion by either the α or the β ray, and that the same proportion of the total energy is used up in producing ions, an approximate estimate can be made of the ratio of the energy 197radiated by the α and β rays by measuring the ratio of the total number of ions produced by them. If λ is the coefficient of absorption of the β rays in air, the rate of production of ions per unit volume at a distance x from the source is
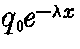
where q₀ is the rate of ionization at the source.
The total number of ions produced by complete absorption of the rays is
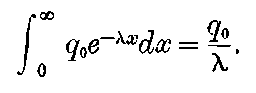
Now λ is difficult to measure experimentally for air, but an approximate estimate can be made of its value from the known fact that the absorption of β rays is approximately proportional to the density of any given substance.
For β rays from uranium the value of λ for aluminium is about 14, and λ divided by the density is 5·4. Taking the density of air as ·0012, we find that for air
λ = ·0065.
The total number of ions produced in air is thus 154q₀ when the rays are completely absorbed.
Now from the above table the ionization due to the β rays is ·0074 of that produced by α rays, when the β rays passed through a distance of 5·7 cms. of air.
Thus we have approximately
Therefore about ⅙ of the total energy radiated into air by a thin layer of uranium is carried by the β rays or electrons. The ratio for thorium is about ¹⁄₂₂ and for radium about ¹⁄₁₄, assuming the rays to have about the same average value of λ.
This calculation takes into account only the energy which is radiated out into the surrounding gas; but on account of the ease with which the α rays are absorbed, even with a thin layer, the greater proportion of the radiation is absorbed by the radio-active substance itself. This is seen to be the case when it is recalled that the α radiation of thorium or radium is reduced to half value after passing through a thickness of about 0·0005 cm. of 198aluminium. Taking into consideration the great density of the radio-active substances, it is probable that most of the radiation which escapes into the air is due to a thin skin of the powder not much more than ·0001 cm. in thickness.
An estimate, however, of the relative rate of emission of energy by the α and β rays from a thick layer of material can be made in the following way:—For simplicity suppose a thick layer of radio-active substance spread uniformly over a large plane area. There seems to be no doubt that the radiations are emitted uniformly from each portion of the mass; consequently, the radiation, which produces the ionizing action in the gas above the radio-active layer, is the sum total of all the radiation which reaches the surface of the layer.
Let λ1 be the average coefficient of absorption of the α rays in the radio-active substance itself and σ the specific gravity of the substance. Let E1 be the total energy radiated per sec. per unit mass of the substance when the absorption of the rays in the substance itself is disregarded. The energy per sec. radiated to the upper surface by a thickness dx of a layer of unit area at a distance x from the surface is given by
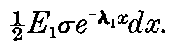
The total energy W1 per unit area radiated to the surface per sec. by a thickness d is given by
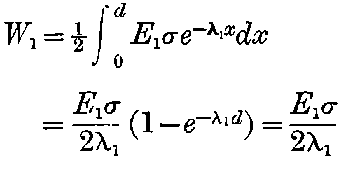
if λ1d is large.
In a similar way it may be shown that the energy W2 of the β rays reaching the surface is given by
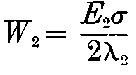
where E2 and λ2 are the values for the β rays corresponding to E1 and λ1 for the α rays. Thus it follows that
199λ1 and λ2 are difficult to determine directly for the radio-active substance itself, but it is probable that the ratio λ1/λ2 is not very different from the ratio for the absorption coefficients for another substance like aluminium. This follows from the general result that the absorption of both α and β rays is proportional to the density of the substance; for it has already been shown in the case of the β rays from uranium that the absorption of the rays in the radio-active material is about the same as for non-radio-active matter of the same density.
With a thick layer of uranium oxide spread over an area of 22 sq. cms., it was found that the saturation current between parallel plates 6·1 cms. apart, due to the α rays, was 12·7 times as great as the current due to the β rays. Since the α rays were entirely absorbed between the plates and the total ionization produced by the β rays is 154 times the value at the surface of the plates,
Now the value of λ1 for aluminium is 2740 and of λ2 for the same metal 14, thus
This shows that the energy radiated from a thick layer of material by the β rays is only about 1 per cent. of the energy radiated in the form of α rays.
This estimate is confirmed by calculations based on independent data. Let m1, m2 be the masses of the α and β particles respectively and v1, v2 their velocities.
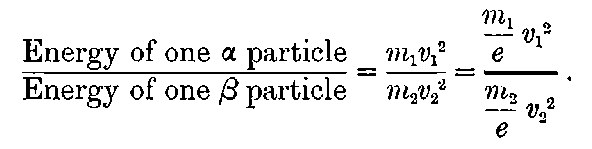
Now it has been shown that for the α rays of radium
200The velocity of the β rays of radium varies between wide limits. Taking for an average value
it follows that the energy of the α particle from radium is almost 83 times the energy of the β particle. If equal numbers of α and β particles are projected per second, the total energy radiated in the form of α rays is about 83 times the amount in the form of β rays.
Evidence will be given later (section 253) to show that the number of α particles projected is probably four times the number of β particles; so that a still greater proportion of the energy is emitted in the form of α rays. These results thus lead to the conclusion that, from the point of view of the energy emitted, the α rays are far more important than the β rays. This conclusion is supported by other evidence which is discussed in chapters XII and XIII, where it will be shown that the α rays play by far the most important part in the changes occurring in radio-active bodies, and that the β rays only appear in the latter stages of the radio-active processes. From data based on the relative absorption and ionization of the β and γ rays in air, it can be shown that the γ rays carry off about the same amount of energy as the β rays. These conclusions are confirmed by direct measurement of the heating effect of radium, which is discussed in detail in chapter XII.
115. Besides their power of acting on a photographic plate, and of ionizing gases, the radiations from active bodies are able to produce marked chemical and physical actions in various substances. Most of these effects are due either to the α or β rays. The γ rays produce little effect in comparison. Since the β rays are similar in all respects to high velocity cathode rays, it is to be expected that they will produce effects similar in character to those produced by the cathode rays in a vacuum tube.
Becquerel[184] has studied the action of radium rays in producing phosphorescence in various bodies. The substance to be tested was placed above the radium in the form of powder on a very thin mica plate. Examination was made of the sulphides of calcium and strontium, ruby, diamond, varieties of spar, phosphorus and hexagonal blende. Substances like the ruby and spar, which phosphoresce under luminous rays, did not phosphoresce under the radium rays. On the other hand, those which were made luminous by ultra-violet light were also luminous under the action of radium rays. The radium rays show distinct differences from X rays. For example, a diamond which was very luminous with radium rays was unaffected by X rays. The double sulphate of uranium and potassium is more luminous than hexagonal blende under X rays, but the reverse is true for radium rays; under the influence of these rays, sulphide of calcium gave a blue luminosity but was hardly affected by X rays.
202The following table shows the relative phosphorescence excited in various bodies.
| Substance | Without screen. Intensity | Across screen of black paper |
|---|---|---|
| Hexagonal blende | 13·36 | ·04 |
| Platino-cyanide of barium | 1·99 | ·05 |
| Diamond | 1·14 | ·01 |
| Double sulphate of Uranium and Potassium | 1·00 | ·31 |
| Calcium Fluoride | ·30 | ·02 |
In the last column the intensity without the screen is in each case taken as unity. The great diminution of intensity after the rays have passed through black paper shows that most of the phosphorescence developed without the screen is, in the majority of cases, due to the α rays.
Bary[185] has made a very complete examination of the class of substances which become luminous under radium rays. He found that the great majority of substances belong to the alkali metals and alkaline earths. All these substances were also phosphorescent under the action of X rays.
Crystalline zinc sulphide (Sidot’s blende) phosphoresces very brightly under the influence of the rays from radium and other very active substances. This was observed by Curie and Debierne in their study of the radium emanation and the excited activity produced by it. It has also been largely used by Giesel as an optical means of detecting the presence of emanations from very active substances. It is an especially sensitive means of detecting the presence of α rays, when it exhibits the “scintillating” property already discussed in section 96. In order to show the luminosity due to the α rays, the screen should be held close to the active substance, as the rays are absorbed in their passage through a few centimetres of air. Zinc sulphide is also luminous under the action of the β rays, but the phosphorescence is far more persistent than when produced by the α rays.
Very beautiful luminous effects are produced by large crystals of the platinocyanides exposed to the radium rays. Those 203containing lithium give a brilliant pink colour. The calcium and barium salts fluoresce with a deep green light, and the sodium compound with a lemon yellow. The mineral willemite (zinc silicate) was recently found by Kunz to be an even more sensitive means of detecting the presence of the radiations than platinocyanide of barium. It fluoresces showing a beautiful greenish colour, and a piece of mineral exposed to the action of the rays appears quite translucent. The crystals of the platinocyanides of barium and lithium are especially suited for showing the action of the γ rays, and, in this respect, are superior to willemite.
A very striking effect is shown by the mineral kunzite—a new variety of spodumene discovered by Kunz[186]. This is a transparent gem like crystal, often of very large size, which glows with a beautiful reddish colour under the action of the β or γ rays, but does not appear to be sensitive to the α rays. The luminosity extends throughout the crystal, but is not so marked as in the platinocyanides or willemite. The mineral sparteite[187], a form of calcite containing a few per cent. of manganese, has been found by Ambrecht to fluoresce with a very deep orange light under the β and γ rays. The colour appears to depend on the intensity of the rays, and is deeper close to the radium than at some distance away.
If kunzite and sparteite are exposed to the action of the cathode rays in a vacuum tube, the colour is different from that produced by the radium rays. The former appears a deep yellow, instead of the deep red observed with the radium rays.
The different actions of the radium rays on these fluorescent substances can be illustrated very simply and beautifully by the following experiment. A small U tube is filled with fragments of the fluorescent substance arranged in layers. The U tube is immersed in liquid air and the emanation from about 30 mgrs. of radium bromide is condensed in the tube. On closing the tube and removing it from the liquid air, the emanation distributes itself uniformly in the tube. The shades of colour produced in the different substances are clearly seen.
It is observed that all the crystals increase in luminosity for several hours, on account of the excited activity produced 204by the emanation. This effect is especially observed in kunzite, which at first hardly responds to the rays, since the β and γ rays, which causes it to fluoresce, are not given out by the emanation itself but by one of its later products. The intensity of the β and γ rays is, in consequence, small at first but rises to a maximum after several hours; the luminosity observed varies in a corresponding manner.
Sir William Crookes[188] has made an examination of the effect of continued exposure of a diamond to the radium rays. An “off-colour” diamond, of a pale yellow colour, was placed inside a tube with radium bromide. After 78 days’ exposure, the diamond had darkened and become bluish green in tint; when heated at 50° in a mixture of potassium chlorate for ten days, the diamond lost its dull surface colour and was bright and transparent, and its tint had changed to a pale bluish green. The rays have thus a double action on the diamond; the less penetrating β rays produce a superficial darkening due to the change of the surface into graphite, while the more penetrating β rays and the γ rays produce a change of colour throughout its mass. The diamond phosphoresced brightly during the whole course of its exposure to the rays. Crookes also observed that the diamond still retained enough activity to affect a photographic plate 35 days after removal, although, during the period of 10 days, it was heated in a mixture sufficiently powerful to remove the outer skin of graphite. This residual activity may possibly be due to a slow transformation product of the emanation which is deposited on the surface of bodies (see chapter XI).
Marckwald observed that the α rays from radio-tellurium produced marked phosphorescence on some kinds of diamonds. An account of the various luminous effects produced on different gems by exposure to the radium and actinium rays has been given by Kunz and Baskerville[189].
Both zinc sulphide and platinocyanide of barium diminish in luminosity after exposure for some time to the action of the rays. To regenerate a screen of the latter, exposure to solar light is necessary. A similar phenomenon has been observed by Villard 205for a screen exposed to Röntgen rays. Giesel made a screen of platinocyanide of radio-active barium. The screen, very luminous at first, gradually turned brown in colour, and at the same time the crystals became dichroic. In this condition the luminosity was much less, although the active substance had increased in activity after preparation. Many of the substances which are luminous under the rays from active substances lose this property to a large extent at low temperatures[190].
116. Luminosity of radium compounds. All radium compounds are spontaneously luminous. This luminosity is especially brilliant in the dry haloid salts, and persists for long intervals of time. In damp air the salts lose a large amount of their luminosity, but they recover it on drying. With very active radium chloride, the Curies have observed that the light changes in colour and intensity with time. The original luminosity is recovered if the salt is dissolved and dried. Many inactive preparations of radiferous barium are strongly luminous. The writer has seen a preparation of impure radium bromide which gave out a light sufficient to read by in a dark room. The luminosity of radium persists over a wide range of temperature and is as bright at the temperature of liquid air as at ordinary temperatures. A slight luminosity is observed in a solution of radium, and if crystals are being formed in the solution, they can be clearly distinguished in the liquid by their greater luminosity.
117. Spectrum of the phosphorescent light of radium and actinium. Compounds of radium, with a large admixture of barium, are usually strongly self-luminous. This luminosity decreases with increasing purity, and pure radium bromide is only very feebly self-luminous. A spectroscopic examination of the slight phosphorescent light of pure radium bromide has been made by Sir William and Lady Huggins[191]. On viewing the light with a direct vision spectroscope, there were faint indications of a variation of luminosity at different points along the spectrum. In 206order to get a photograph of the spectrum within a reasonable time, they made use of a quartz spectroscope of special design which had been previously employed in a spectroscopic examination of faint celestial objects. After three days’ exposure with a slit of ¹⁄₄₅₀ of an inch in width, a negative was obtained which showed a number of bright lines. The magnified spectrum is shown in Fig. 46 A. The lines of this spectrum were found to agree not only in position but also in relative intensity with the band spectrum of nitrogen. The band spectrum of nitrogen and also the spark spectrum[192] of radium are shown in the same figure.
Some time afterwards Sir William Crookes and Prof. Dewar showed that this spectrum of nitrogen was not obtained if the radium was contained in a highly exhausted tube. Thus it appears that the spectrum is due to the action of the radium rays either on occluded nitrogen or the nitrogen in the atmosphere surrounding the radium.
It is very remarkable that a phosphorescent light, like that of radium bromide, should show a bright line spectrum of nitrogen. It shows that radium at ordinary temperatures is able to set up radiations which are produced only by the electric discharge under special conditions.
Sir William and Lady Huggins were led to examine the spectrum of the natural phosphorescent light of radium with the hope that some indications might be obtained thereby of the processes occurring in the radium atom. Since the main radiation from radium consists of positively charged atoms projected with great velocity, radiations must be set up both in the expelled body and in the system from which it escapes.

Fig. 46a.
Giesel[193] observed that the spectrum of the phosphorescent light of actinium consists of three bright lines. Measurements of the wave length were made by Hartmann[194]. The luminosity was very slight and a long exposure was required. The lines observed were 207in the red, blue and green. The wave length λ and velocity are shown below.
| Line | Intensity | λ |
| 1 | 10 | 4885·4 ± 0·1 Ångström units |
| 2 | 6 | 5300 ± 6 „ |
| 3 | 1 | 5909 ± 10 „ |
The line 4885 was very broad; the other two lines were so feeble that it was difficult to determine their wave length with accuracy. Hartmann suggests that these lines may be found in the spectrum of the new stars. The lines observed have no connection with radium or its emanation[195].
118. Thermo-luminescence. E. Wiedemann and Schmidt[196] have shown that certain bodies after exposure to the cathode rays or the electric spark become luminous when they are heated to a temperature much below that required to cause incandescence. This property of thermo-luminescence is most strikingly exhibited in certain cases where two salts, one of which is much in excess of the other, are precipitated together. It is to be expected that such bodies would also acquire the property when exposed to the β or cathodic rays of radium. This has been found to be the case by Wiedemann[197]. Becquerel showed that fluor-spar, exposed to the radium rays, was luminous when heated. The glass tubes in which radium is kept are rapidly blackened. On heating the tube, a strong luminosity is observed, and the coloration to a large extent disappears. The peculiarity of many of these bodies lies in the fact that the property of becoming luminous when heated is retained for a long interval of time after the body is removed from the influence of the exciting cause. It appears probable that the rays cause chemical changes in these bodies, which are permanent until heat is applied. A portion of the chemical energy is then released in the form of visible light.
119. Some electric effects. Radium rays have the same effect as ultra-violet light and Röntgen rays in increasing the 208facility with which a spark passes between electrodes. Elster and Geitel[198] showed that if two electrodes were separated by a distance such that the spark just refused to pass, on bringing near a specimen of radium the spark at once passes. This effect is best shown with short sparks from a small induction coil. The Curies have observed that radium completely enveloped by a lead screen 1 cm. thick produces a similar action. The effect in that case is due to the γ rays alone. This action of the rays can be very simply illustrated by connecting two spark-gaps with the induction coil in parallel. The spark-gap of one circuit is adjusted so that the discharge just refuses to pass across it, but passes by the other. When some radium is brought near the silent spark-gap, the spark at once passes and ceases in the other[199].
Hemptinne[200] found that the electrodeless discharge in a vacuum tube began at a higher pressure when a strong preparation of radium was brought near the tube. In one experiment the discharge without the rays began at 51 mms. but with the radium rays at 68 mms. The colour of the discharge was also altered.
Himstedt[201] found that the resistance of selenium was diminished by the action of radium rays in the same way as by ordinary light.
F. Henning[202] examined the electrical resistance of a barium chloride solution containing radium of activity 1000, but could observe no appreciable difference between it and a similar pure solution of barium chloride. This experiment shows that the action of the rays from the radium does not produce any appreciable change in the conductivity of the barium solution.
Kohlrausch and Henning[203] have recently made a detailed examination of the conductivity of pure radium bromide solutions, and have obtained results very similar to those for the corresponding barium solutions. Kohlrausch[204] found that the conductivity of water exposed to the radiations from radium increased more rapidly than water which had not been exposed. 209This increase of conductivity may have been due to an increase of the conductivity of the water itself, or to an increased rate of solution of the glass of the containing vessel.
Specimens of strongly active material have been employed to obtain the potential at any point of the atmosphere. The ionization due to the active substance is so intense that the body to which it is attached rapidly takes up the potential of the air surrounding the active substance. In this respect it is more convenient and rapid in its action than the ordinary taper or water dropper, but on account of the disturbance of the electric field by the strong ionization produced, it is probably not so accurate a method as that of the water dropper.
120. Effect on liquid and solid dielectrics. P. Curie[205] made the very important observation that liquid dielectrics became partial conductors under the influence of radium rays. In these experiments the radium, contained in a glass tube, was placed in an inner thin cylinder of copper. This was surrounded by a concentric copper cylinder, and the liquid to be examined filled the space between. A strong electric field was applied, and the current through the liquid measured by means of an electrometer.
The following numbers illustrate the results obtained:
| Substance | Conductivity in megohms per 1 cm.3 |
|---|---|
| Carbon bisulphide | 20 × 10-14 |
| Petroleum ether | 15 „ |
| Amyline | 14 „ |
| Carbon chloride | 8 „ |
| Benzene | 4 „ |
| Liquid air | 1·3 „ |
| Vaseline oil | 1·6 „ |
Liquid air, vaseline oil, petroleum ether, amyline, are normally nearly perfect insulators. The conductivity of amyline and petroleum ether due to the rays at -17° C. was only ⅒ of its value at 0° C. There is thus a marked action of temperature on the conductivity. For very active material the current was 210proportional to the voltage. With material of only ¹⁄₅₀₀ of the activity, it was found that Ohm’s law was not obeyed.
The following numbers were obtained:
| Volts | Current |
|---|---|
| 50 | 109 |
| 100 | 185 |
| 200 | 255 |
| 400 | 335 |
For an increase of voltage of 8 times, the current only increases about 3 times. The current in the liquid thus tends to become “saturated” as does the ordinary ionization current through a gas. These results have an important bearing on the ionization theory, and show that the radiation probably produces ions in the liquid as well as in the gas. It was also found that X rays increased the conductivity to about the same extent as the radium rays.
Becquerel[206] has recently shown that solid paraffin exposed to the β and γ rays of radium acquires the property of conducting electricity to a slight extent. After removal of the radium the conductivity diminishes with time according to the same law as for an ionized gas. These results show that a solid as well as a liquid and gaseous dielectric is ionized under the influence of radium rays.
121. Effect of temperature on the radiations. Becquerel[207], by the electric method, determined the activity of uranium at the temperature of liquid air, and found that it did not differ more than 1 per cent. from the activity at ordinary temperatures. In his experiments, the α rays from the uranium were absorbed before reaching the testing vessel, and the electric current measured was due to the β rays alone. P. Curie[208] found that the luminosity of radium and its power of exciting fluorescence in bodies were retained at the temperature of liquid air. Observations by the electric method showed that the activity of radium was unaltered at the temperature of liquid air. If a radium compound is heated in an open vessel, it is found that the activity, measured by the α rays, falls to about 25 per cent. of its original value. This is however not due to a change in the radio-activity, but to the release of the radio-active emanation, which is stored in the 211radium. No alteration is observed if the radium is heated in a closed vessel from which none of the radio-active products are able to escape.
122. Motion of radium in an electric field. Joly[209] found that a disc, one side of which is coated with a few milligrams of radium bromide, exhibits, when an electrified body is brought near it, motions very different to those observed in the case of an inactive substance. The electrified body, whether positive or negative, repels the suspended body if brought up to it on the side coated with radium, but attracts it if presented to the naked side.
This effect is very simply shown by constructing a small apparatus like a radiometer. Two covered glasses are attached to the end of a glass fibre about 6 cms. long, the surfaces lying in the same plane. The apparatus is free to rotate on a pivot. The two vanes are coated on alternate faces with radium bromide, and the whole apparatus contained within a glass receiver. If an electrified rod of ebonite or sealing wax is brought up close to the receiver, a rotation is communicated to the vane which increases as the pressure of the air is lowered to 5 or 6 cms. of mercury. By placing the apparatus between parallel plates connected with the terminals of a Wimshurst machine, a steady rotation is communicated to the vanes. The rotation is always in such a direction that the radium coated surface is repelled from the electrified body.
This action was examined still further by attaching the vanes to the glass beam of a Coulomb’s balance. A metal sphere, which could be charged from without, was fixed facing the side coated with radium. A repulsion was always observed except when the charge was very strong and the vane near the sphere. If, however, the two vanes were connected by a light wire and a similar sphere placed exactly opposite the other, an attraction was observed if one sphere was charged, but a repulsion if both were charged. These effects were observed whether the vanes were of aluminium or glass.
Joly found that the effect could not be explained by any direct 212action due to the movement of the ions in an electric field. The recoil, due to the expulsion of α particles from one side of the vane, is far too small to account for the movement observed.
This effect can, I think, be simply accounted for by taking into consideration the difference in conductivity of the gas on the two sides of the radium coated vane. If a small vane, coated uniformly with radium on both sides, and mounted on an insulating support, be brought near a charged body kept at a constant potential, it acts like a water dropper and rapidly acquires very nearly the average potential which existed at that point before the vane was brought up. The mechanical force acting on the vane will, in consequence, be small. If, however, the vane is only coated with radium on the side near the charged body, the ionization and consequently the conductivity of the gas is much greater between the vane and the charged body than on the opposite side. Suppose, for simplicity, the body is charged to a positive potential. On account of the greater conductivity of the gas on the side facing the charged body, it will rapidly acquire a positive charge, and the potential of the vane will reach a higher value than existed at that place before the vane was introduced. This will result in a repulsion of the vane. This also accounts for the attraction observed in the experiment with the Coulomb’s balance already referred to. Suppose that one sphere is positively charged and the other earthed, and the two vanes metallically connected together. The vane next to the charged body will become charged positively, but this charge will be dissipated rapidly on account of the ionization of the gas close to the opposite vane, and, in most conditions, this loss of charge will be so rapid that the potential of the vane is unable to reach the value which would exist at that place in the field, if the vane were removed. There will, in consequence, be an attracting force acting on the vane towards the sphere.
The repulsion observed by Joly is thus only an indirect result of the ionization in the gas produced by the radium, and should be shown under conditions where similar unequal distribution of ionization is produced by any other sources.
Since radium gives out heat at a fairly rapid rate, a radiometer in which the vanes were coated on one side with radium instead of lampblack, should rotate at low pressure of the gas, even if no 213source of light is brought near it. This should evidently be the case, since the face coated with radium should reach a slightly higher temperature than the other. This experiment has been tried, but the effect seems too small to produce rotation of the vanes.
123. Rays from active radium preparations change oxygen into ozone[210]. Its presence can be detected by the smell or by the action on iodide of potassium paper. This effect is due to the α and β rays from the radium, and not to the luminous rays from it. Since energy is required to produce ozone from oxygen, this must be derived from the energy of the radiations.
The Curies found that radium compounds rapidly produced coloration in glass. For moderately active material the colour is violet, for more active material it is yellow. Long continued action blackens the glass, although the glass may have no lead in its composition. This coloration gradually extends through the glass, and is dependent to some extent on the kind of glass used.
Giesel[211] found that he could obtain as much coloration in rock-salt and fluor-spar by radium rays, as by exposure to the action of cathode rays in a vacuum tube. The coloration, however, extended much deeper than that produced by the cathode rays. This is to be expected, since the radium rays have a higher velocity, and consequently greater penetrating power, than the cathode rays produced in an ordinary vacuum tube. Goldstein observed that the coloration is far more intense and rapid when the salts are melted or heated to a red heat. Melted potassium sulphate, under the action of a very active preparation of radium, was rapidly coloured a strong greenish blue which gradually changed into a dark green. Salomonsen and Dreyer[212] found that plates of quartz were coloured by exposure to radium rays. When examined minutely, plates cut perpendicular to the optic axis showed the presence of lines and striae, parallel to the binary axes. Adjacent portions of the striated system differed considerably in intensity of 214coloration and clearly revealed the heterogeneity of structures of the crystal.
The cause of these colorations by cathode and radium rays has been the subject of much discussion. Elster and Geitel[213] observed that a specimen of potassium sulphate, coloured green by radium rays, showed a strong photo-electric action, i.e. it rapidly lost a negative charge of electricity when exposed to the action of ultra-violet light. All substances coloured by cathode rays show a strong photo-electric action, and, since the metals sodium and potassium themselves show photo-electric action to a very remarkable degree, Elster and Geitel have suggested that the colorations are caused by a solid solution of the metal in the salt.
Although the coloration due to radium rays extends deeper than that due to the cathode rays, when exposed to light the colour fades away at about the same rate in the two cases.
Becquerel[214] found that white phosphorus is changed into the red variety by the action of radium rays. This action was shown to be due mainly to the β rays. The secondary radiation set up by the primary rays also produced a marked effect. Radium rays, like ordinary light rays, also caused a precipitate of calomel in the presence of oxalic acid.
Hardy and Miss Wilcock[215] found that a solution of iodoform in chloroform turned purple after exposure for 5 minutes to the rays from 5 milligrams of radium bromide. This action is due to the liberation of iodine. By testing the effect of screens of different thicknesses, over the radium, this action was found to be mainly due to the β rays from the radium. Röntgen rays produce a similar coloration.
Hardy[216] also observed an action of the radium rays on the coagulation of globulin. Two solutions of globulin from ox serum were used, one made electro-positive by adding acetic acid, and the other electro-negative by adding ammonia. When the globulin was exposed close to the radium in naked drops, the opalescence of the electro-positive solution rapidly diminished, showing that the 215solution became more complete. The electro-negative solution was rapidly turned to a jelly and became opaque. These actions were found to be due to the α rays of radium alone.
This is further evidence in favour of the view that the α rays consist of projected positively charged bodies of atomic dimensions, for a similar coagulation effect is produced by the metallic ions of liquid electrolytes, and has been shown by W. C. D. Whetham[217] to be due to the electric charges carried by the ions.
124. Gases evolved from radium. Curie and Debierne[218] observed that radium preparations placed in a vacuum tube continually lowered the vacuum. The gas evolved was always accompanied by the emanation, but no new lines were observed in its spectrum. Giesel[219] has observed a similar evolution of gas from solutions of radium bromide. Giesel forwarded some active material to Runge and Bödlander, in order that they might test the gas spectroscopically. From 1 gram of a 5 per cent. radium preparation they obtained 3·5 c.c. of gas in 16 days. This gas was found, however, to be mainly hydrogen, with 12 per cent. of oxygen. In later experiments Ramsay and Soddy[220] found that 50 milligrams of radium bromide evolved gases at the rate of about 0·5 c.c. per day. This is a rate of evolution about twice that observed by Runge and Bödlander. On analysing the gases about 28·9 per cent. consisted of oxygen, and the rest hydrogen. The slight excess of hydrogen over that attained in the decomposition of water, they consider to be due to the action of oxygen on the grease of the stop-cocks. The radio-active emanation from radium has a strong oxidizing action and rapidly produces carbon dioxide, if carbonaceous matter is present. The production of gas is probably due to the action of the radiations in decomposing water. The amount of energy required to produce the rate of decomposition observed by Ramsay and Soddy—about 10 c.c. per day for 1 gram of radium bromide—corresponds to about 30 gram-calories per day. This amount of energy is about two per cent. of the total energy emitted in the form of heat.
216Ramsay and Soddy (loc. cit.) have also observed the presence of helium in the gases evolved by solution of radium bromide. This important result is considered in detail in section 267.
125. Walkhoff first observed that radium rays produce burns of much the same character as those caused by Röntgen rays. Experiments in this direction have been made by Giesel, Curie and Becquerel, and others, with very similar results. There is at first a painful irritation, then inflammation sets in, which lasts from 10 to 20 days. This effect is produced by all preparations of radium, and appears to be due mainly to the α and β rays.
Care has to be taken in handling radium on account of the painful inflammation set up by the rays. If a finger is held for some minutes at the base of a capsule containing a radium preparation, the skin becomes inflamed for about 15 days and then peels off. The painful feeling does not disappear for two months.
Danysz[221] found that this action is mainly confined to the skin, and does not extend to the underlying tissue. Caterpillars subjected to the action of the rays lost their power of motion in several days and finally died.
Radium rays have been found beneficial in certain cases of cancer. The effect is apparently similar to that produced by Röntgen rays, but the use of radium possesses the great advantage that the radiating source can be enclosed in a fine tube and introduced at the particular point at which the action of the rays is required. The rays have also been found to hinder or stop the development of microbes[222].
It would be out of place here to give an account of the numerous experiments that have been made by physicists and physiologists on the action of the rays of radium and of other radio-active substances on different organisms, such as caterpillars, mice and guinea-pigs. In some cases, the experiments have been carried out by placing the organisms in an atmosphere impregnated 217with the radium emanation. The effect of an exposure under such conditions for several days or weeks has been found generally harmful and in many cases fatal. The literature in this new department of study is already large and is increasing rapidly.
Another interesting action of the radium rays has been observed by Giesel. On bringing up a radium preparation to the closed eye, in a dark room, a sensation of diffuse light is observed. This effect has been examined by Himstedt and Nagel[223] who have shown that it is due to a fluorescence produced by the rays in the eye itself. The blind are able to perceive this luminosity if the retina is intact, but not if the retina is diseased. Hardy and Anderson[224] have examined this effect in some detail. The sensation of light is produced both by the β and γ rays. The eyelid practically absorbs all the β rays, so that the luminosity observed with a closed eye is due to the γ rays alone. The lens and retina of the eye are strongly phosphorescent under the action of the β and γ rays. Hardy and Anderson consider that the luminosity observed in a dark room with the open eye (the phosphorescent light of the radium itself being stopped by black paper) is to a large extent due to the phosphorescence set up in the eyeball. The γ rays, for the most part, produce the sensation of light when they strike the retina.
Tommasina stated that the air exhaled by man contained a larger proportion of ions than ordinary air, and, in consequence, caused an increased rate of discharge of an electroscope. The experiment was repeated by Elster and Geitel but with negative results. On the other hand, they found that the breath of Dr Giesel, of Braunschweig, who had been engaged continuously in the chemical separation of the radio-active bodies, caused a rapid loss of charge of an electroscope. This increased rate of discharge was probably mainly due to the radium emanation, with which his system had become impregnated by inhaling the emanation-laden air of the laboratory.
126. An account will now be given of some experiments which have thrown much light, not only on the nature of the processes which serve to maintain the radio-activity of the radio-active bodies, but also on the source of the energy continuously emitted by those bodies. In this chapter, for simplicity, the radio-activity of uranium and thorium will alone be considered, for it will be seen later that the changes taking place in these two substances are typical of those which occur in all radio-active substances.
We have seen (section 23) that there is some doubt whether the radio-activity of thorium is due to that element itself, or to an unknown radio-active constituent associated with it. This uncertainty, however, will present no serious difficulty when we are discussing the radio-activity of thorium, for the general conclusions are, for the most part, independent of whether thorium is the primary radio-active constituent or not. For simplicity, however, it will be assumed for the present that the radio-activity is due to thorium itself. If future research should definitely show that the radio-activity, ordinarily observed in thorium, is due to a new radio-active element mixed with it, the radio-active processes considered will refer to this new element.
127. Uranium X. The experiments of Mme Curie show that the radio-activity of uranium and radium is an atomic phenomenon. The activity of any uranium compound depends only on the amount of that element present, and is unaffected by its chemical combination with other substances, and is not appreciably affected by wide variations of temperature. It would thus seem 219probable, since the activity of uranium is a specific property of the element, that the activity could not be separated from it by chemical agencies.
In 1900, however, Sir William Crookes[225] showed that, by a single chemical operation, uranium could be obtained photographically inactive while the whole of the activity could be concentrated in a small residue free from uranium. This residue, to which he gave the name of Ur X, was many hundred times more active photographically, weight for weight, than the uranium from which it had been separated. The method employed for this separation was to precipitate a solution of the uranium with ammonium carbonate. On dissolving the precipitate in an excess of the reagent, a light precipitate remained behind. This was filtered, and constituted the Ur X. The active substance Ur X was probably present in very small quantity, mixed with impurities derived from the uranium. No new lines were observed in its spectrum. A partial separation of the activity of uranium was also effected by another method. Crystallized uranium nitrate was dissolved in ether, when it was found that the uranium divided itself between the ether and water present in two unequal fractions. The small part dissolved in the water layer was found to contain practically all the activity when examined by the photographic method, while the other fraction was almost inactive. These results, taken by themselves, pointed very strongly to the conclusion that the activity of uranium was not due to the element itself, but to some other substance, associated with it, which had distinct chemical properties.
Results of a similar character were observed by Becquerel[226]. It was found that barium could be made photographically very active by adding barium chloride to the uranium solution and precipitating the barium as sulphate. By a succession of precipitations the uranium was rendered photographically almost inactive, while the barium was strongly active.
The inactive uranium and the active barium were laid aside; but, on examining them a year later, it was found that the uranium had completely regained its activity, while that of the barium had 220completely disappeared. The loss of activity of uranium was thus only temporary in character.
In the above experiments, the activity of uranium was examined by the photographic method. The photographic action produced by uranium is due almost entirely to the β rays. The α rays, in comparison, have little if any effect. Now the radiation from Ur X consists entirely of β rays, and is consequently photographically very active. If the activity of uranium had been measured electrically without any screen over it, the current observed would have been due very largely to the α rays, and little change would have been observed after the removal of Ur X, since only the constituent responsible for the β rays was removed. This important point is discussed in more detail in section 205.
128. Thorium X. Rutherford and Soddy[227], working with thorium compounds, found that an intensely active constituent could be separated from thorium by a single chemical operation. If ammonia is added to a thorium solution, the thorium is precipitated, but a large amount of the activity is left behind in the filtrate, which is chemically free from thorium. This filtrate was evaporated to dryness, and the ammonium salts driven off by ignition. A small residue was obtained which, weight for weight, was in some cases several thousand times more active than the thorium from which it was obtained, while the activity of the precipitated thorium was reduced to less than one half of its original value. This active constituent was named Th X from analogy to Crookes’ Ur X.
The active residue was found to consist mainly of impurities from the thorium; the Th X could not be examined chemically, and probably was present only in minute quantity. It was also found that an active constituent could be partly separated from thorium oxide by shaking it with water for some time. On filtering the water, and evaporating down, a very active residue was obtained which was analogous in all respects to Th X.
On examining the products a month later, it was found that the Th X was no longer active, while the thorium had completely 221regained its activity. A long series of measurements was then undertaken to examine the time-rate of these processes of decay and recovery of activity.
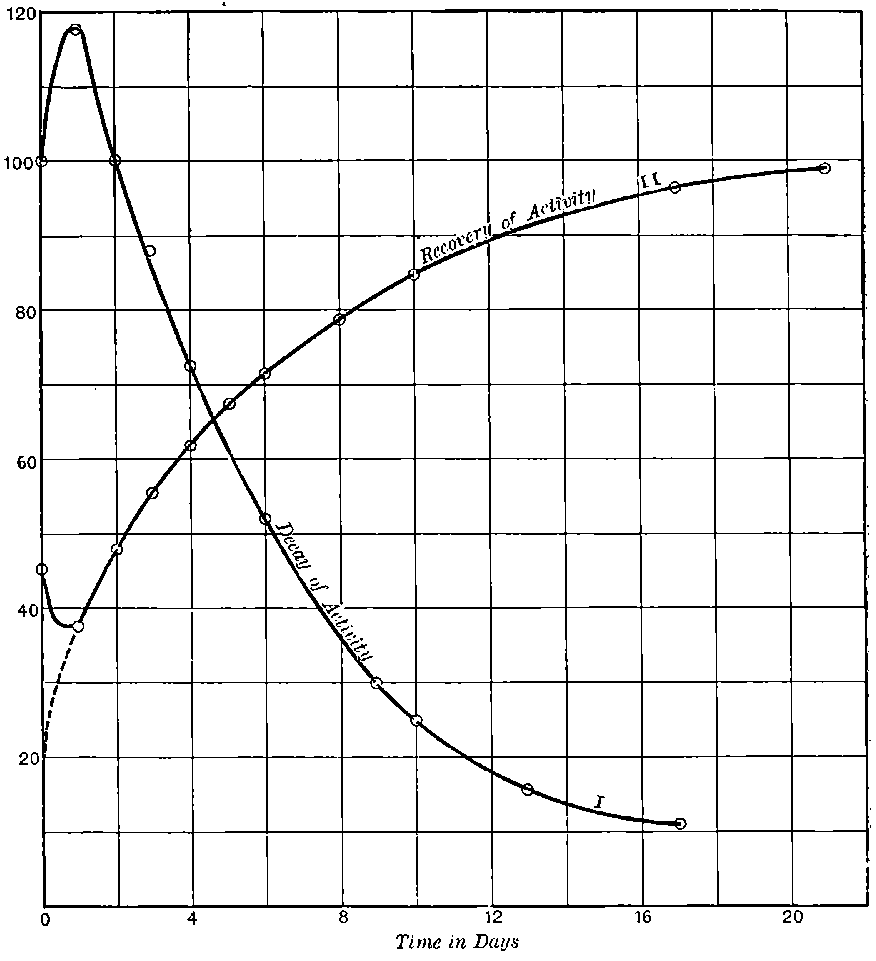
Fig. 47.
The results are shown graphically in Fig. 47, where the final activity of the thorium and the initial activity of the Th X are in each case taken as 100. The ordinates represent the activities determined by means of the ionization current, and the abscissae represent the time in days. It will be observed that both curves are irregular for the first two days. The activity of the Th X increased at first, while the activity of the thorium diminished. Disregarding these initial irregularities of the curves, which will be 222explained in detail in section 208, it will be seen that, after the first two days, the time taken for the thorium to recover half its lost activity is about equal to the time taken by the Th X to lose half its activity. This time in each case is about four days. The percentage proportion of the activity regained by the thorium, over any given interval, is approximately equal to the percentage proportion of the activity lost by the Th X during the same interval.
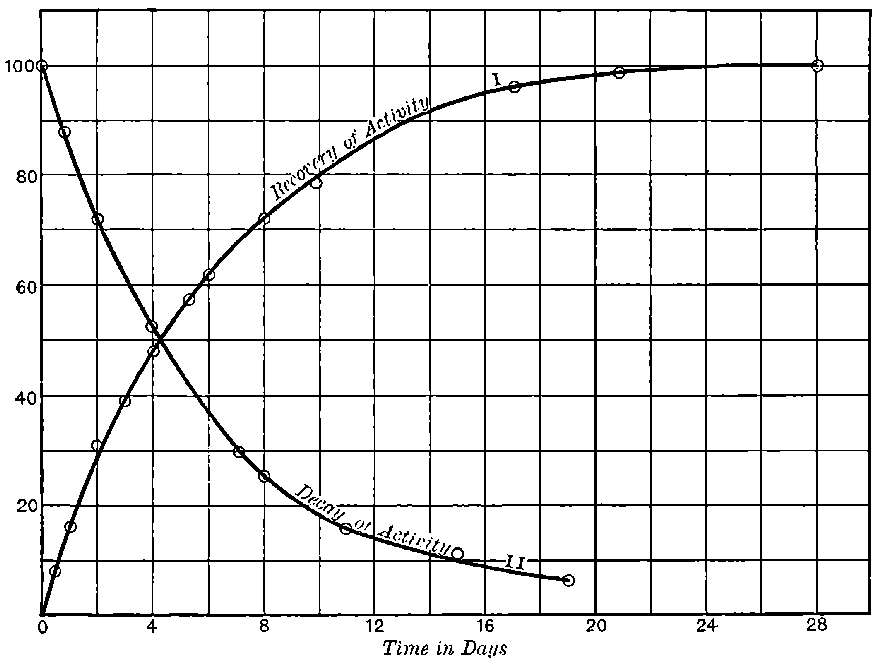
Fig. 48.
If the recovery curve is produced backwards to meet the vertical axis, it does so at a minimum of 25 per cent., and the above conclusions hold more accurately, if the recovery is assumed to start from this minimum. This is clearly shown by Fig. 48, where the percentages of activity recovered, reckoned from the 25 per cent. minimum, are plotted as ordinates. In the same figure the decay curve, after the second day, is shown on the same scale. The activity of the Th X decays with the time according to an exponential law, falling to half value in about four days. If I₀ is the initial activity and It is the activity after a time t, then
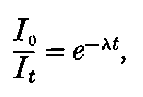
223where λ is a constant and e the natural base of logarithms. The experimental curve of the rise of activity from a minimum to a maximum value is therefore expressed by the equation
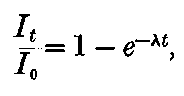
where I₀ is the amount of activity recovered when the state of constant activity is reached, It the activity recovered after a time t, and λ is the same constant as before.
129. Uranium X. Similar results were obtained when uranium was examined. The Ur X was separated by Becquerel’s method of successive precipitations with barium. The decay of the separated activity and the recovery of the lost activity are shown graphically in Fig. 49. A more detailed discussion of this experiment is given in section 205.
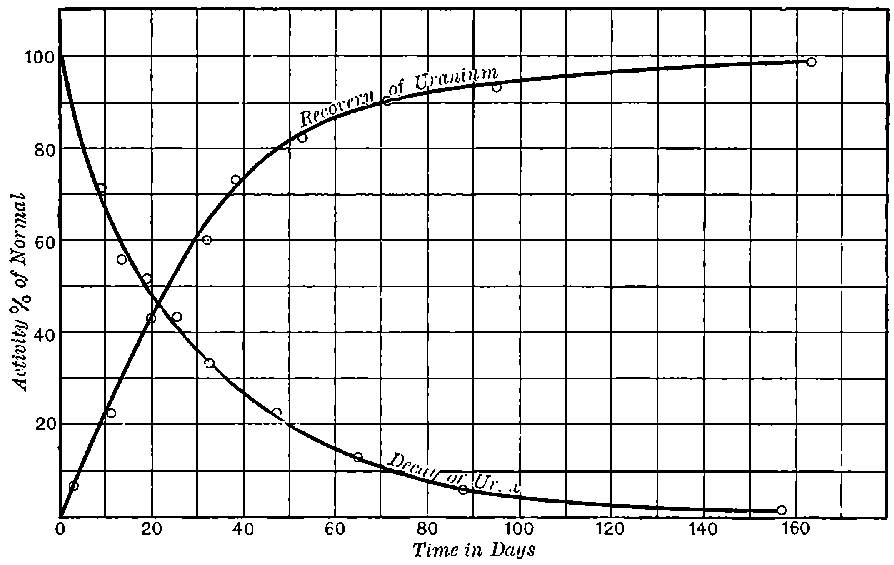
Fig. 49.
The curves of decay and recovery exhibit the same peculiarities and can be expressed by the same equations as in the case of thorium. The time-rate of decay and recovery is, however, much slower than for thorium, the activity of the Ur X falling to half its value in about 22 days.
224A large number of results of a similar character have been obtained from other radio-active products, separated from the radio-elements, but the cases of thorium and uranium will suffice for the present to form a basis for the discussion of the processes that are taking place in radio-active bodies.
130. Theory of the phenomena. These processes of decay and recovery go on at exactly the same rate if the substances are removed from the neighbourhood of one another, or enclosed in lead, or placed in a vacuum tube. It is at first sight a remarkable phenomenon that the processes of decay and recovery should be so intimately connected, although there is no possibility of mutual interaction between them. These results, however, receive a complete explanation on the following hypotheses:
(1) That there is a constant rate of production of fresh radio-active matter by the radio-active body;
(2) That the activity of the matter so formed decreases according to an exponential law with the time from the moment of its formation.
Suppose that q₀ particles of new matter are produced per second from a given mass of matter. The rate of emission of energy due to the particles produced in the time dt, is, at the moment of their formation, equal to Kq₀dt, where K is a constant.
It is required to find the activity due to the whole matter produced after the process has continued for a time T.
The activity dI, due to the matter produced during the time dt at the time t, decays according to an exponential law during the time T – t that elapses before its activity is estimated, and in consequence is given by
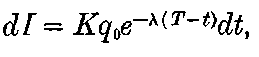
where λ is the constant of decay of activity of the active matter. The activity IT due to the whole matter produced in the time T is thus given by
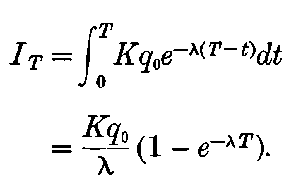
225The activity reaches a maximum value I₀ when T is very great, and is then given by
thus
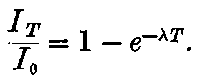
This equation agrees with the experimental results for the recovery of lost activity. Another method for obtaining this equation is given later in section 133.
A state of equilibrium is reached when the rate of loss of activity of the matter already produced is balanced by the activity supplied by the production of new active matter. According to this view, the radio-active bodies are undergoing change, but the activity remains constant owing to the action of two opposing processes. Now, if this active matter can at any time be separated from the substance in which it is produced, the decay of its activity, as a whole, should follow an exponential law with the time, since each portion of the matter decreases in activity according to an exponential law with the time, whatever its age may be. If I₀ is the initial activity of the separated product, the activity It after an interval t is given by
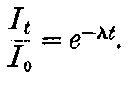
Thus, the two assumptions—of uniform production of active matter and of the decay of its activity in an exponential law from the moment of its formation—satisfactorily explain the relation between the curves of decay and recovery of activity.
131. Experimental evidence. It now remains to consider further experimental evidence in support of these hypotheses. The primary conception is that the radio-active bodies are able to produce from themselves matter of chemical properties different from those of the parent substance, and that this process goes on at a constant rate. This new matter initially possesses the property of activity, and loses it according to a definite law. The fact that a proportion of the activity of radium and thorium can be concentrated in small amounts of active matter like Th X 226or Ur X does not, of itself, prove directly that a material constituent responsible for the activity has been chemically separated. For example, in the case of the separation of Th X from thorium, it might be supposed that the non-thorium part of the solution is rendered temporarily active by its association with thorium, and that this property is retained through the processes of precipitation, evaporation, and ignition, and finally manifests itself in the residue remaining. According to this view it is to be expected that any precipitate capable of removing the thorium completely from its solution should yield active residues similar to those obtained from ammonia. No such case has, however, been observed. For example, when thorium nitrate is precipitated by sodium or ammonium carbonate, the residue from the filtrate after evaporation and ignition is free from activity and the thorium carbonate obtained has the normal amount of activity. In fact, ammonia is the only reagent yet found capable of completely separating Th X from thorium. A partial separation of the Th X can be made by shaking thorium oxide with water owing to the greater solubility of Th X in water.
Thorium and uranium behave quite differently with regard to the action of ammonia and ammonium carbonate. Ur X is completely precipitated with the uranium in an ammonia solution and the filtrate is inactive. Ur X is separated by ammonium carbonate, while Th X under the same conditions is completely precipitated with the thorium. The Ur X and the Th X thus behave like distinct types of matter with well-marked chemical properties quite distinct from those of the substances in which they are produced. The removal of Ur X by the precipitation of barium is probably not directly connected with the chemical properties of Ur X. The separation is probably due to the dragging down of the Ur X with the dense barium precipitate. Sir William Crookes found that the Ur X was dragged down by precipitates when no question of insolubility was involved, and such a result is to be expected if the Ur X exists in extremely minute quantity. It must be borne in mind that the actual amount of the active constituents Th X and Ur X, separated from thorium and uranium, is probably infinitesimal, and that the greater proportion of the residues is due to impurities present 227in the salt and the reagents, a very small amount of active matter being mixed with them.
132. Rate of production of Th X. If the recovery of the activity of uranium or thorium is due to the continuous production of new active matter, it should be possible to obtain experimental evidence of the process. As the case of thorium has been most fully investigated, a brief account will be given of some experiments made by Rutherford and Soddy[228] to show that Th X is produced continuously at a constant rate. Preliminary experiments showed that three successive precipitations were sufficient to remove the Th X almost completely from the thorium. The general method employed was to precipitate a solution of 5 grams of thorium-nitrate with ammonia. The precipitate was then redissolved in nitric acid and the thorium again precipitated as before, as rapidly as possible, so that the Th X produced in the time between successive precipitations should not appreciably affect the results. The removal of the Th X was followed by measurements of the activity of the residues obtained from successive filtrates. In three successive precipitations the activities of the residues were proportional to 100, 8, 1·6 respectively. Thus two precipitations are nearly sufficient to free the thorium from Th X.
The thorium freed from Th X was then allowed to stand for a definite time, and the amount of Th X formed during that time found by precipitating it, and measuring its radio-activity. According to the theory, the activity It of the thorium formed in the time t is given by
where I₀ is the total activity of Th X, when there is radio-active equilibrium.
If λt is small,
Since the activity of Th X falls to half value in 4 days, the 228value of λ expressed in hours = ·0072. After standing a period of 1 hour about ¹⁄₁₄₀, after 1 day ⅙, after 4 days ½ of the maximum should be obtained. The experimental results obtained showed an agreement, as good as could be expected, with the equation expressing the result that the Th X was being produced at a constant rate.
The thorium-nitrate which had been freed from Th X was allowed to stand for one month, and then it was again subjected to the same process. The activity of the Th X was found to be the same as that obtained from an equal amount of the original thorium-nitrate. In one month, therefore, the Th X had been regenerated, and had reached a maximum value. By leaving the thorium time to recover fully its activity, this process can be repeated indefinitely, and equal amounts of Th X are obtained at each precipitation. Ordinary commercial thorium-nitrate and the purest nitrate obtainable showed exactly the same action, and equal amounts of Th X could be obtained from equal weights. These processes thus appear to be independent of the chemical purity of the substance[229].
The process of the production of Th X is continuous, and no alteration has been observed in the amount produced in the given time after repeated separations. After 23 precipitations extending over 9 days, the amount produced in a given interval was about the same as at the beginning of the process.
These results are all in agreement with the view that the Th X is being continuously produced from the thorium compound at a constant rate. The amount of active matter produced from 1 gram of thorium is probably extremely minute, but the electrical effects due to its activity are so large that the process of production can be followed after extremely short intervals. With a sensitive electrometer the amount of Th X produced per minute in 10 grams of thorium-nitrate gives a rapid movement to the electrometer needle. For larger intervals it is necessary to add additional capacity to the system to bring the effects within range of the instrument.
229 133. Rate of decay of activity. It has been shown that the activity of Ur X and Th X decays according to an exponential law with the time. This, we shall see later, is the general law of decay of activity in any type of active matter, obtained by itself, and freed from any secondary active products which it may, itself, produce. In any case, when this law is not fulfilled, it can be shown that the activity is due to the superposition of two or more effects, each of which decays in an exponential law with the time. The physical interpretation of this law still remains to be discussed.
It has been shown that in uranium and thorium compounds there is a continuous production of active matter which keeps the compound in radio-active equilibrium. The changes by which the active matter is produced must be chemical in nature, since the products of the action are different in chemical properties from the matter in which the changes take place. The activity of the products has afforded the means of following the changes occurring in them. It now remains to consider the connection between the activity at any time, and the amount of chemical change taking place at that time.
In the first place, it is found experimentally that the saturation ionization current it, after the active product has been allowed to decay for a time t, is given by
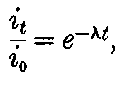
where i₀ is the initial saturation current and λ the constant of decay.
Now the saturation current is a measure of the total number of ions produced per second in the testing vessel. It has already been shown that the α rays, which produce the greater proportion of ionization in the gas, consist of positively charged particles projected with great velocity. Suppose for simplicity that each atom of active matter, in the course of its change, gives rise to one projected α particle. Each α particle will produce a certain average number of ions in its path before it strikes the boundaries or is absorbed in the gas. Since the number of projected particles per second is equal to the number of atoms changing per second, 230the number of atoms nt which change per second at the time t is given by
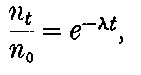
where n₀ is the initial number which change per second. On this view, then, the law of decay expresses the result that the number of atoms changing in unit time, diminishes according to an exponential law with the time. The number of atoms Nt which remain unchanged after an interval t is given by
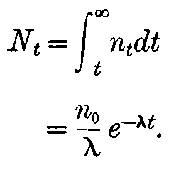
If N₀ is the number of atoms at the beginning,
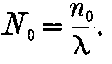
Thus
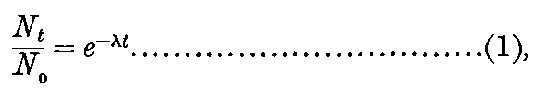
or the law of decay expresses the fact that the activity of a product at any time is proportional to the number of atoms which remain unchanged at that time.
This is the same as the law of monomolecular change in chemistry, and expresses the fact that there is only one changing system. If the change depended on the mutual action of two systems, the law of decay would be different, since the rate of decay in that case would depend on the relative concentration of the two reacting substances. This is not so, for not a single case has yet been observed in which the law of decay was affected by the amount of active matter present.
From the above equation (1)
or the number of systems changing in unit time is proportional to the number unchanged at that time.
In the case of recovery of activity, after an active product has been removed, the number of systems changing in unit time, when 231radio-active equilibrium is produced, is equal to λN₀. This must be equal to the number q₀ of new systems applied in unit time, or
λ has thus a distinct physical meaning, and may be defined as the proportion of the total number of systems present which change per second. It has different values for different types of active matter, but is invariable for any particular type of matter. For this reason, λ will be termed the “radio-active constant„ of the product.
We are now in a position to discuss with more physical definiteness the gradual growth of Th X in thorium, after the Th X has been completely removed from it. Let q₀ particles of Th X be produced per second by the thorium, and let N be the number of particles of Th X present at any time t after the original Th X was removed. The number of particles of Th X which change every second is λN, where λ is the radio-active constant of Th X. Now, at any time during the process of recovery, the rate of increase of the number of particles of Th X = the rate of production – the rate of change; that is
The solution of this equation is of the form
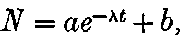
where a and b are constants.
Now when t is very great, the number of particles of Th X present reach a maximum value N₀.
and the equation becomes
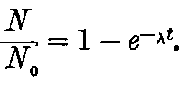
This is equivalent to the equation already obtained in section 130, 232since the intensity of the radiation is always proportional to the number of particles present.
134. Influence of conditions on the rate of decay. Since the activity of any product, at any time, may be taken as a measure of the rate at which chemical change takes place, it may be used as a means of determining the effect of conditions on the changes occurring in radio-active matter. If the rate of change should be accelerated or retarded, it is to be expected that the value of the radio-active constant λ will be increased or decreased, i.e. that the decay curve will be different under different conditions.
No such effect, however, has yet been observed in any case of radio-active change, where none of the active products produced are allowed to escape from the system. The rate of decay is unaltered by any chemical or physical agency, and in this respect the changes in radio-active matter are sharply distinguished from ordinary chemical changes. For example, the rate of decay of activity from any product takes place at the same rate when the substance is exposed to light as when it is kept in the dark, and at the same rate in a vacuum as in air or any other gas at atmospheric pressure. Its rate of decay is unaltered by surrounding the active matter by a thick layer of lead under conditions where no ordinary radiation from outside can affect it. The activity of the matter is unaffected by ignition or chemical treatment. The material giving rise to the activity can be dissolved in acid and re-obtained by evaporation of the solution without altering the activity. The rate of decay is the same whether the active matter is retained in the solid state or kept in solution. When a product has lost its activity, resolution or heat does not regenerate it, and as we shall see later, the rate of decay of the active products, so far examined, is the same at a red heat as at the temperature of liquid air. In fact, no variation of physical or chemical conditions has led to any observable difference in the decay of activity of any of the numerous types of active matter which have been examined.
135. Effect of conditions on the rate of recovery of activity. The recovery of the activity of a radio-element with 233time, when an active product is separated from it, is governed by the rate of production of fresh active matter and by the decay of activity of that already produced. Since the rate of decay of the activity of the separated product is independent of conditions, the rate of recovery of activity can be modified only by a change of the rate of production of fresh active matter. As far as experiments have gone, the rate of production, like the rate of decay, is independent of chemical or physical conditions. There are indeed certain cases which are apparent exceptions to this rule. For example, the escape of the radio-active emanations from thorium and radium is readily affected by heat, moisture and solution. A more thorough investigation, however, shows that the exception is only apparent and not real. These cases will be discussed more in detail in chapter VII, but it may be stated here that the differences observed are due to differences in the rate of escape of the emanations into the surrounding gas, and not to differences in the rate of production. For this reason it is difficult to test the question at issue in the case of the thorium compounds, which in most cases readily allow the emanation produced by them to escape into the air.
In order to show that the rate of production is independent of molecular state, temperature, etc., it is necessary in such a case to undertake a long series of measurements extending over the whole time of recovery. It is impossible to make accurate relative comparisons to see if the activity is altered by the conversion of one compound into another. The relative activity in such a case, when measured by spreading a definite weight of material uniformly on a metal plate, varies greatly with the physical conditions of the precipitate, although the total activity of two compounds may be the same.
The following method[230] offers an accurate and simple means of studying whether the rate of production of active matter is influenced by molecular state. The substance is chemically converted into any compound required, care being taken that active products are recovered during the process. The new compound is then spread on a metal plate and compared with a standard sample of uranium for several days or weeks as required. If the rate of 234production of active matter is altered by the conversion, there should be an increase or decrease of activity to a new steady value, where the production of active matter is again balanced by the rate of decay. This method has the great advantage of being independent of the physical condition of the precipitate. It can be applied satisfactorily to a compound of thorium like the nitrate and the oxide which has been heated to a white heat, after which treatment only a slight amount of emanation escapes. The nitrate was converted into the oxide in a platinum crucible by treatment with sulphuric acid and ignition to a white heat. The oxide so obtained was spread on a plate, but no change of its activity was observed with time, showing that in this case the rate of production was independent of molecular state. This method, which is limited in the case of thorium, may be applied generally to the uranium compounds where the results are not complicated by the presence of an emanation.
No differences have yet been observed in the recovery curves of different thorium compounds after the removal of Th X. For example, the rate of recovery is the same whether the precipitated hydroxide is converted into the oxide or into the sulphate.
136. Disintegration hypothesis. In the discussion of the changes in radio-active bodies, only the active products Ur X and Th X have been considered. It will, however, be shown later that these two products are only examples of many other types of active matter which are produced by the radio-elements, and that each of these types of active matter has definite chemical as well as radio-active properties, which distinguish it, not only from the other active products, but also from the substance from which it is produced.
The full investigation of these changes will be shown to verify in every particular the hypothesis that radio-activity is the accompaniment of chemical changes of a special kind occurring in matter, and that the constant activity of the radio-elements is due to an equilibrium process, in which the rate of production of fresh active matter balances the rate of change of that already formed.
The nature of the process taking place in the radio-elements, 235in order to give rise to the production at a constant rate of new kinds of active matter, will now be considered. Since in thorium or uranium compounds there is a continuous production of radio-active matter, which differs in chemical properties from the parent substance, some kind of change must be taking place in the radio-element. This change, by which new matter is produced, is very different in character from the molecular changes dealt with in chemistry, for no chemical change is known which proceeds at the same rate at the temperatures corresponding to a red heat and to liquid air, and is independent of all physical and chemical actions. If, however, the production of active matter is supposed to be the result of changes, not in the molecule, but in the atom itself, it is not to be expected that the temperature would exert much influence. The general experience of chemistry in failing to transform the elements by the action of temperature is itself strong evidence that wide ranges of temperature have not much effect in altering the stability of the chemical atom.
The view that the atoms of the radio-elements are undergoing spontaneous disintegration was put forward by Rutherford and Soddy as a result of evidence of this character. The discovery of the material nature of the α rays added strong confirmation to the hypothesis; for it has been pointed out (section 95) that the expulsion of α particles must be the result of a disintegration of the atoms of the radio-element. Taking the case of thorium as an example, the processes occurring in the atom may be pictured in the following way. It must be supposed that the thorium atoms are not permanently stable systems, but, on an average, a constant small proportion of them—about one atom in every 1016 will suffice—break up per second. The disintegration consists in the expulsion from the atom of one or more α particles with great velocity. For simplicity, it will be supposed that each atom expels one α particle. It has been shown that the α particle of radium has a mass about twice that of the hydrogen atom. From the similarity of the α rays from thorium and radium, it is probable that the α particle of thorium does not differ much in mass from that of radium, and may be equal to it. The α particles expelled from the thorium atoms as they break up constitute what is known as the “non-separable activity” of thorium. This activity, 236measured by the α rays, is about 25 per cent. of the maximum. After the escape of an α particle, the part of the atom left behind, which has a mass slightly less than that of the thorium atom, tends to rearrange its components to form a temporarily stable system. It is to be expected that it will differ in chemical properties from the thorium atom from which it was derived. The atom of the substance Th X is, on this view, the thorium atom minus one α particle. The atoms of Th X are far more unstable than the atoms of thorium, and one after the other they break up, each atom expelling one α particle as before. These projected α particles give rise to the radiation from the Th X. Since the activity of Th X falls to half its original value in about four days, on an average half of the atoms of Th X break up in four days, the number breaking up per second being always proportional to the number present. After an atom of Th X has expelled an α particle, the mass of the system is again reduced, and its chemical properties are changed. It will be shown (section 154) that the Th X produces the thorium emanation, which exists as a radio-active gas, and that this in turn is transformed into matter which is deposited on solid bodies and gives rise to the phenomena of excited activity. The first few successive changes occurring in thorium are shown diagrammatically below (Fig. 50).
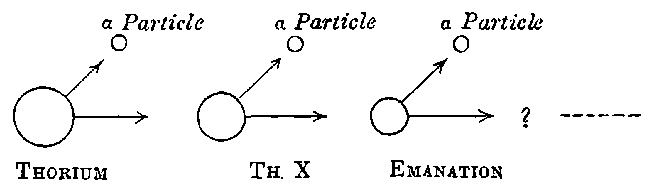
Fig. 50.
Thus as a result of the disintegration of the thorium atom, a series of chemical substances is produced, each of which has distinctive chemical properties. Each of these products is radio-active, and loses its activity according to a definite law. Since thorium has an atomic weight of 237, and the weight of the α particle is about 2, it is evident that, if only one α particle is expelled at each change, the process of disintegration could pass through a number of successive stages and yet leave behind, 237at the end of the process, a mass comparable with that of the parent atom.
It will be shown later that a process of disintegration, very similar to that already described for thorium, must be supposed to take place also in uranium, actinium and radium. The full discussion of this subject cannot be given with advantage until two of the most important products of the three substances thorium, radium and actinium, viz. the radio-active emanations and the matter which causes excited activity, have been considered in detail.
137. Magnitude of the changes. It can be calculated by several independent methods (see section 246) that, in order to account for the radio-activity observed in thorium, about 3 × 104 atoms in each gram of thorium suffer disintegration per second. It is well known (section 39) that 1 cubic centimetre of hydrogen at atmospheric pressure and temperature contains about 3·6 × 1019 molecules. From this it follows that one gram of thorium contains 3·6 × 1021 atoms. The fraction which breaks up per second is thus about 10-17. This is an extremely small ratio, and it is evident that the process could continue for long intervals of time, before the amount of matter changed would be capable of detection by the spectroscope or by the balance. With the electroscope it is possible to detect the radiation from 10-5 gram of thorium, i.e. the electroscope is capable of detecting the ionization which accompanies the disintegration of a single thorium atom per second. The electroscope is thus an extraordinarily delicate means for detection of minute changes in matter, which are accompanied, as in the case of the radio-elements, by the expulsion of charged particles with great velocity. It is possible to detect by its radiation the amount of Th X produced in a second from 1 gram of thorium, although the process would probably need to continue thousands of years before it could be detected by the balance or the spectroscope. It is thus evident that the changes occurring in thorium are of an order of magnitude quite different from that of ordinary chemical changes, and it is not surprising that they have never been observed by direct chemical methods.
138. Introduction. A most important and striking property possessed by radium, thorium, and actinium, but not by uranium or polonium, is the power of continuously emitting into the surrounding space a material emanation, which has all the properties of a radio-active gas. This emanation is able to diffuse rapidly through gases and through porous substances, and may be separated from the gas with which it is mixed by condensation by the action of extreme cold. This emanation forms a connecting link between the activity of the radio-elements themselves and their power of exciting activity on surrounding objects, and has been studied more closely than the other active products on account of its existence in the gaseous state. The emanations from the three active bodies all possess similar radio-active properties, but the effects are more marked in the case of the emanation from radium, on account of the very great activity of that element.
139. Discovery of the emanation. In the course of examination of the radiations of thorium, several observers had noted that some of the thorium compounds, and especially the oxide, were very inconstant sources of radiation, when examined in open vessels by the electrical method. Owens[231] found that this inconstancy was due to the presence of air currents. When a closed vessel was used, the current, immediately after the introduction of the active matter, increased with the time, and finally 239reached a constant value. By drawing a steady stream of air through the vessel the value of the current was much reduced. It was also observed that the radiations could apparently pass through large thicknesses of paper, which completely absorbed the ordinary α radiation.
In an investigation of these peculiar properties of thorium compounds, the writer[232] found that the effects were due to an emission of radio-active particles of some kind from the thorium compounds. This “emanation,” as it was termed for convenience, possesses the properties of ionizing the gas and acting on a photographic plate, and is able to diffuse rapidly through porous substances like paper and thin metal foil.
The emanation, like a gas, is completely prevented from escaping by covering the active matter with a thin plate of mica. The emanation can be carried away by a current of air; it passes through a plug of cotton-wool and can be bubbled through solutions without any loss of activity. In these respects, it behaves very differently from the ions produced in the gas by the rays from active substances, for these give up their charges completely under the same conditions.
Since the emanation passes readily through large thicknesses of cardboard, and through filters of tightly packed cotton-wool, it does not seem likely that the emanation consists of particles of dust given off by the active matter. This point was tested still further by the method used by Aitken and Wilson, for detecting the presence of dust particles in the air. The oxide, enclosed in a paper cylinder, was placed in a glass vessel, and the dust was removed by repeated small expansions of the air over a water surface. The dust particles act as nuclei for the formation of small drops and are then removed from the air by the action of gravity. After repeated expansions, no cloud was formed, and the dust was considered to be removed. After waiting for some time to allow the thorium emanation to collect, further expansions were made but no cloud resulted, showing that for the small expansions used, the particles were too small to become centres of condensation. The emanation then could not be regarded as dust emitted from thorium.
240Since the power of diffusing rapidly through porous substances, and acting on a photographic plate, is also possessed by a chemical substance like hydrogen peroxide, some experiments were made to see if the emanation could be an agent of that character. It was found, however, that hydrogen peroxide is not radio-active, and that its action on the plate is a purely chemical one, while it is the radiation from the emanation and not the emanation itself that produces ionizing and photographic effects.
140. Experimental arrangements. The emanation from thorium is given off in minute quantity. No appreciable lowering of the vacuum is observed when an emanating compound is placed in a vacuum tube and no new spectrum lines are observed.
For an examination of the emanation, an apparatus similar in principle to that shown in Fig. 51 is convenient.
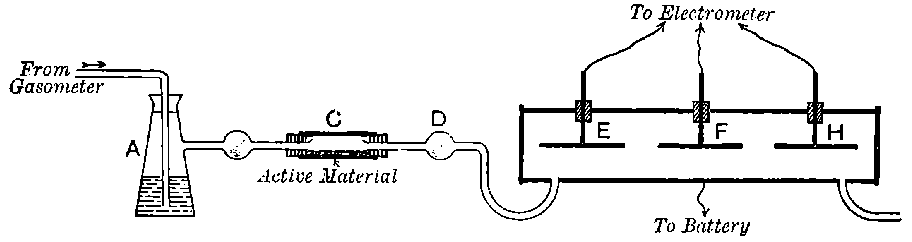
Fig. 51.
The thorium compound, either bare or enclosed in a paper envelope, was placed in a glass tube C. A current of air from a gasometer, after passing through a tube containing cotton-wool to remove dust particles, bubbled through sulphuric acid in the vessel A. It then passed through a bulb containing tightly packed cotton-wool to prevent any spray being carried over. The emanation, mixed with air, was carried from the vessel C through a plug of cotton-wool D, which removed completely all the ions carried with the emanation. The latter then passed into a long brass cylinder, 75 cm. in length and 6 cm. in diameter. The insulated cylinder was connected with a battery in the usual way. Three insulated electrodes, E, F, H, of equal lengths, were placed along the axis of the cylinder, supported by brass rods passing through ebonite corks in the side of the cylinder. The current through the gas, due to the presence of the emanation, was measured by means of 241an electrometer. An insulating key was arranged so that any one of the electrodes E, F, H could be rapidly connected with one pair of quadrants of the electrometer, the other two being always connected with earth. The current observed in the testing cylinder vessel was due entirely to the ions produced by the emanation carried into the vessel by the current of air. On substituting a uranium compound for the thorium, not the slightest current was observed. After a constant flow has passed for about 10 minutes, the current due to the emanation reaches a constant value.
The variation of the ionization current with the voltage is similar to that observed for the gas ionized by the radiations from the active bodies. The current at first increases with the voltage, but finally reaches a saturation value.
141. Duration of the activity of the emanation. The emanation rapidly loses its activity with time. This is very readily shown with the apparatus of Fig. 51. The current is found to diminish progressively along the cylinder, and the variation from electrode to electrode depends on the velocity of the flow of air.
If the velocity of the air current is known, the decay of activity of the emanation with time can be deduced. If the flow of air is stopped, and the openings of the cylinder closed, the current steadily diminishes with time. The following numbers illustrate the variation with time of the saturation current, due to the emanation in a closed vessel. The observations were taken successively, and as rapidly as possible after the current of air was stopped.
| Time in seconds | Current |
| 0 | 100 |
| 28 | 69 |
| 62 | 51 |
| 118 | 25 |
| 155 | 14 |
| 210 | 6·7 |
| 272 | 4·1 |
| 360 | 1·8 |
Curve A, Fig. 52, shows the relation existing between the current through the gas and the time. The current just before the flow of air was stopped is taken as unity. The current through 242the gas, which is a measure of the activity of the emanation, diminishes according to an exponential law with the time like the activity of the products Ur X and Th X. The rate of decay is, however, much more rapid, the activity of the emanation decreasing to half value in about one minute. According to the view developed in section 136, this implies that half of the emanation particles have undergone change in one minute. After an interval of 10 minutes the current due to the emanation is very small, showing that practically all the emanation particles present have undergone change.
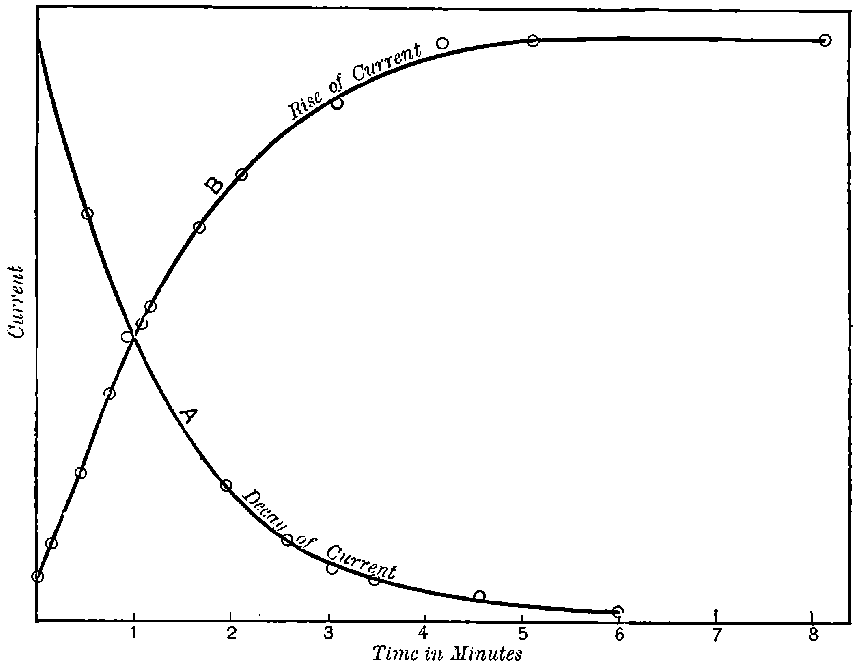
Fig. 52.
The rate of decay has been more accurately determined by Rossignol and Gimingham[233] who found that the activity fell to half value in about 51 seconds. Bronson[234], using the steady deflection method described in section 69, found the corresponding time 54 seconds.
The decrease of the current with the time is an actual measure of the decrease of the activity of the emanation, and is not in any 243way influenced by the time that the ions produced take to reach the electrodes. If the ions had been produced from a uranium compound the duration of the conductivity for a saturation voltage would only have been a fraction of a second.
The rate of decay of the activity of the emanation is independent of the electromotive force acting on the gas. This shows that the radio-active particles are not destroyed by the electric field. The current through the gas at any particular instant, after stoppage of the flow of air, was found to be the same whether the electromotive force had been acting the whole time or had been just applied for the time of the test.
The emanation itself is unaffected by a strong electric field and so cannot be charged. By testing its activity after passing it through long concentric cylinders, charged to a high potential, it was found that the emanation certainly did not move with a velocity greater than ·00001 cm. per second, for a gradient of 1 volt per cm., and there was no evidence to show that it moved at all. This conclusion has been confirmed by the experiments of McClelland[235].
The rate at which the emanation is produced is independent of the gas surrounding the active matter. If in the apparatus of Fig. 51 air is replaced by hydrogen, oxygen, or carbonic acid, similar results are obtained, though the current observed in the testing vessel varies for the different gases on account of the unequal absorption by them of the radiation from the emanation.
If a thorium compound, enclosed in paper to absorb the α radiation, is placed in a closed vessel, the saturation current due to the emanation is found to vary directly as the pressure. Since the rate of ionization is proportional to the pressure for a constant source of radiation, this experiment shows that the rate of emission of the emanation is independent of the pressure of the gas. The effect of pressure on the rate of production of the emanation is discussed in more detail later in section 157.
142. Effect of thickness of layer. The amount of emanation emitted by a given area of thorium compound depends on the thickness of the layer. With a very thin layer, the current between two parallel plates, placed in a closed vessel as in Fig. 17, is due very largely to the α rays. Since the α radiation is very 244readily absorbed, the current due to it practically reaches a maximum when the surface of the plate is completely covered by a thin layer of the active material. On the other hand the current produced by the emanation increases until the layer is several millimetres in thickness, and then is not much altered by adding fresh active matter. This falling off of the current after a certain thickness has been reached is to be expected, since the emanation, which takes several minutes to diffuse through the layer above it, has already lost a large proportion of its activity.
With a thick layer of thorium oxide in a closed vessel, the current between the plates is largely due to the radiation from the emanation lying between the plates. The following tables illustrate the way in which the current varies with the thickness of paper for both a thin and a thick layer.
Table I. Thin Layer.
Thickness of sheets of paper ·0027.
| No. of layers of paper | Current |
|---|---|
| 0 | 1 |
| 1 | ·37 |
| 2 | ·16 |
| 3 | ·08 |
Table II. Thick Layer.
Thickness of paper ·008 cm.
| No. of layers of paper | Current |
|---|---|
| 0 | 1 |
| 1 | ·74 |
| 2 | ·74 |
| 5 | ·72 |
| 10 | ·67 |
| 20 | ·55 |
The initial current with the unscreened compound is taken as unity. In Table I, for a thin layer of thorium oxide, the current diminished rapidly with additional layers of thin paper. In this case the current is due almost entirely to the α rays. In Table II the current falls to ·74 for the first layer. In this case about 26% of the current is due to the α rays, which are practically absorbed by the layer ·008 cm. in thickness. The slow decrease with additional layers shows that the emanation diffuses so rapidly through a few layers of paper that there is little loss of activity during the passage. The time taken to diffuse through 20 layers is however appreciable, and the current consequently has decreased. After passing through a layer of cardboard 1·6 mms. in thickness the current is reduced to about one-fifth of its original value. In 245closed vessels the proportion of the total current, due to the emanation, varies with the distance between the plates as well as with the thickness of the layer of active material. It also varies greatly with the compound examined. In the nitrate, which gives off only a small amount of emanation, the proportion is very much smaller than in the hydroxide, which gives off a large amount of emanation.
143. Increase of current with time. The current due to the emanation does not reach its final value for some time after the active matter has been introduced into the closed vessel. The variation with time is shown in the following table. The saturation current due to thorium oxide, covered with paper, was observed between concentric cylinders of 5·5 cms. and ·8 cm. diameter.
Immediately before observations on the current were made, a rapid stream of air was blown through the apparatus. This removed most of the emanation. However, the current due to the ionization of the gas by the emanation, as it was carried along by the current of air, was still appreciable. The current consequently does not start from zero.
| Time in seconds | Current |
| 0 | 9 |
| 23 | 25 |
| 53 | 49 |
| 96 | 67 |
| 125 | 76 |
| 194 | 88 |
| 244 | 98 |
| 304 | 99 |
| 484 | 100 |
The results are shown graphically in Fig. 52, curve B. The decay of the activity of the emanation with time, and the rate of increase of the activity due to the emanation in a closed space, are connected in the same way as the decay and recovery curves of Th X and Ur X.
With the previous notation, the decay curve is given by
and the recovery curve by
where λ is the radio-active constant of the emanation.
This relation is to be expected, since the decay and recovery 246curves of the emanation are determined by exactly the same conditions as the decay and recovery curves of Ur X and Th X. In both cases there is:
(1) A supply of fresh radio-active particles produced at a constant rate.
(2) A loss of activity of the particles following an exponential law with the time.
In the case of Ur X and Th X, the active matter produced manifests its activity in the position in which it is formed; in this new phenomenon, a proportion of the active matter in the form of the emanation escapes into the surrounding gas. The activity of the emanation, due to a thorium compound kept in a closed vessel, thus reaches a maximum when the rate of supply of fresh emanation particles from the compound is balanced by the rate of change of those already present. The time for recovery of half the final activity is about 1 minute, the same as the time taken for the emanation, when left to itself, to lose half its activity.
If q₀ is the number of emanation particles escaping into the gas per second, and N₀ the final number when radio-active equilibrium is reached, then (section 133),
Since the activity of the emanation falls to half value in 1 minute
and N₀ = 87q₀, or the number of emanation particles present when a steady state is reached is 87 times the number produced per second.
144. Discovery of the emanation. Shortly after the discovery of the thorium emanation, Dorn[236] repeated the results, and, in addition, showed that radium compounds also gave off radio-active emanations, and that the amount given off was much increased by heating the compound. The radium emanation differs from the thorium emanation in the rate at which it loses its activity. It decays far more slowly, but in other respects the emanations of thorium and radium have much the same properties. Both emanations ionize the gas with which they are mixed, and 247affect a photographic plate. Both diffuse readily through porous substances but are unable to pass through a thin plate of mica; both behave like a temporarily radio-active gas, mixed in minute quantity with the air or other gas in which they are conveyed.
145. Decay of activity of the emanation. Very little emanation escapes from radium chloride in the solid state, but the amount is largely increased by heating, or by dissolving the compound in water. By bubbling air through a radium chloride solution, or passing air over a heated radium compound, a large amount of emanation may be obtained which can be collected, mixed with air, in a suitable vessel.
Experiments to determine accurately the rate of decay of activity of the emanation have been made by P. Curie[237], and Rutherford and Soddy[238]. In the experiments of the latter, the emanation mixed with air was stored over mercury in an ordinary gas-holder. From time to time, equal quantities of air mixed with the emanation were measured off by a gas pipette and delivered into a testing vessel. The latter consisted of an air-tight brass cylinder carrying a central insulated electrode. A saturation voltage was applied to the cylinder, and the inner electrode was connected to the electrometer with a suitable capacity in parallel. The saturation current was observed immediately after the introduction of the active gas into the testing vessel, and was taken as a measure of the activity of the emanation present. The current increased rapidly with the time owing to the production of excited activity on the walls of the containing vessel. This effect is described in detail in chapter VIII.
The measurements were made at suitable intervals over a period of 33 days. The following table expresses the results, the initial activity being taken as 100.
| Time in hours | Relative Activity |
|---|---|
| 0 | 100 |
| 20·8 | 85·7 |
| 187·6 | 24·0 |
| 354·9 | 6·9 |
| 521·9 | 1·5 |
| 786·9 | 0·19 |
248The activity falls off according to an exponential law with the time, and decays to half value in 3·71 days. With the usual notation
the mean value of λ deduced from the results is given by
P. Curie determined the rate of decay of activity of the emanation by another method. The active matter was placed at one end of a sealed tube. After sufficient time had elapsed the portion of the tube containing the radium compound was removed. The loss of activity of the emanation, stored in the other part, was tested at regular intervals by observing the ionization current due to the rays which passed through the walls of the glass vessel. The testing apparatus and the connections are shown clearly in Fig. 53. The ionization current is observed between the vessels BB and CC. The glass tube A contains the emanation.
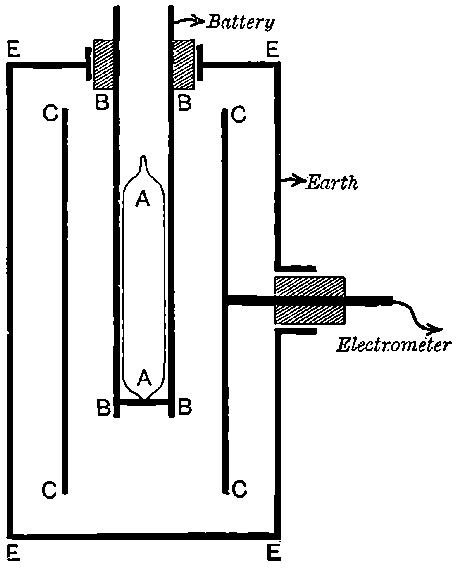
Fig. 53.
Now it will be shown later that the emanation itself gives off only α rays, and these rays are completely absorbed by the glass envelope, unless it is made extremely thin. The rays producing ionization in the testing vessel were thus not due to the α rays from the emanation at all, but to the β and γ rays due to the excited activity produced on the walls of the glass tube by the emanation inside it. What was actually measured was thus the decay of the excited activity derived from the emanation, and not the decay of activity of the emanation itself. Since, however, when a steady state is reached, the amount of excited activity is nearly proportional at any time to the activity of the emanation, the rate of decay of the excited 249activity on the walls of the vessel indirectly furnishes a measure of the rate of decay of the emanation itself. This is only true if the emanation is placed for four or five hours in the tube before observations begin, in order to allow the excited activity time to reach a maximum value.
Using this method P. Curie obtained results similar to those obtained by Rutherford and Soddy by the direct method. The activity decayed according to an exponential law with the time, falling to half value in 3·99 days.
The experiments were performed under the most varied conditions but the rate of decay was found to remain unaltered. The rate of decay did not depend on the material of the vessel containing the emanation or on the nature or pressure of the gas with which the emanation was mixed. It was unaffected by the amount of emanation present, or by the time of exposure to the radium, provided sufficient time had elapsed to allow the excited activity to reach a maximum value before the observations were begun. P. Curie[239] found that the rate of decay of activity was not altered by exposing the vessel containing the emanation to different temperatures, ranging from +450° to -180° C.
In this respect the emanations of thorium and radium are quite analogous. The rate of decay seems to be unaffected by any physical or chemical agency, and the emanations behave in exactly the same way as the radio-active products Th X and Ur X, already referred to. The radio-active constant λ is thus a fixed and unalterable quantity for both emanations, although in one case its value is about 5000 times greater than in the other.
146. Debierne[240] found that actinium gives out an emanation similar to the emanation of thorium and radium. The loss of activity of the emanation is even more rapid than for the thorium emanation, for its activity falls to half value in 3·9 seconds. In consequence of the rapid decay of activity, the emanation is able to diffuse through the air only a short distance from the active matter before it loses the greater proportion of its 250activity. Giesel early observed that the radio-active substance separated by him, which we have seen (section 18) is identical in radio-active properties with actinium, gave off a large amount of emanation. It was in consequence of this property, that he gave it the name of the “emanating substance” and later “emanium.” The impure preparations of this substance emit the emanation very freely and in this respect differ from most of the thorium compounds. The emanation from actinium like those from thorium and radium possesses the property of exciting activity on inactive bodies, but it has not yet been studied so completely as the better known emanations of thorium and radium.
147. With very active specimens of radium a large amount of emanation can be obtained, and the electrical, photographic, and fluorescent effects are correspondingly intense. On account of the small activity of thorium and the rapid decay of its emanation the effects due to it are weak, and can be studied only for a few minutes after its production. The emanation from radium, on the other hand, in consequence of the slow decay of its activity, may be stored mixed with air in an ordinary gas-holder, and its photographic and electrical actions may be examined several days or even weeks after, quite apart from those of the radium from which it was obtained.
It is, in general, difficult to study the radiation due to the emanation alone, on account of the fact that the emanation is continually producing a secondary type of activity on the surface of the vessel in which the emanation is enclosed. This excited activity reaches a maximum value several hours after the introduction of the emanation, and, as long as it is kept in the vessel, this excited activity on the walls decays at the same rate as the emanation itself, i.e. it falls to half its initial value in about 4 days. If, however, the emanation is blown out, the excited activity remains behind on the surface, but rapidly loses its activity in the course of a few hours. After several hours the intensity of the residual radiation is very small.
These effects and their connection with the emanation are discussed more fully in chapter VIII. 251Giesel[241] has recorded some interesting observations of the effect of the radium emanation on a screen of phosphorescent zinc sulphide. When a few centigrams of moist radium bromide were placed on a screen any slight motion of the air caused the luminosity to move to and fro on the screen. The direction of phosphorescence could be altered at will by a slow current of air. The effect was still further increased by placing the active material in a tube and blowing the air through it towards the screen. A screen of barium platinocyanide or of Balmain’s paint failed to give any visible light under the same conditions. The luminosity was not altered by a magnetic field, but it was affected by an electric field. If the screen were charged the luminosity was more marked when it was negative than when it was positive.
Giesel states that the luminosity was not equally distributed, but was concentrated in a peculiar ring-shaped manner over the surface of the screen. The concentration of luminosity on the negative, rather than on the positive, electrode is probably due to the excited activity, caused by the emanation, and not to the emanation itself, for this excited activity is concentrated chiefly on the negative electrode in an electric field (see chapter VIII).
An experiment to illustrate the phosphorescence produced in some substances by the rays from a large amount of emanation is described in section 165.
148. Curie and Debierne[242] have investigated the emanation from radium, and the excited activity produced by it. Some experiments were made on the amount of emanation given off from radium under very low pressures. The tube containing the emanation was exhausted to a good vacuum by a mercury pump. It was observed that a gas was given off from the radium which produced excited activity on the glass walls. This gas was extremely active, and rapidly affected a photographic plate through the glass. It caused fluorescence on the surface of the glass and rapidly blackened it, and was still active after standing ten days. When spectroscopically examined, this gas did not show any new lines, but generally those of the spectra of carbonic acid, hydrogen, 252and mercury. In the light of the results described in section 124 the gas, given off by the radium, was probably the non-active gases hydrogen and oxygen, in which the active emanation was mixed in minute quantity. It will be shown later (section 242) that the energy radiated from the emanation is enormous compared with the amount of matter involved, and that the effects observed, in most cases, are produced by an almost infinitesimal amount of the emanation.
In further experiments, Curie and Debierne[243] found that many substances were phosphorescent under the action of the emanation and the excited activity produced by it. In their experiments, two glass bulbs A and B (Fig. 54) were connected with a glass tube. The active material was placed in the bulb A and the substance to be examined in the other.

Fig. 54.
They found that, in general, substances that were phosphorescent in ordinary light became luminous. The sulphide of zinc was especially brilliant and became as luminous as if exposed to a strong light. After sufficient time had elapsed the luminosity reached a constant value. The phosphorescence is partly due to the excited activity produced by the emanation on its surface, and partly to the direct radiation from the emanation.
Phosphorescence was also produced in glass. Thuringian glass showed the most marked effects. The luminosity of the glass was found to be about the same in the two bulbs, but was more marked in the connecting tube. The effect in the two bulbs was the same even if connected by a very narrow tube.
Some experiments were also made with a series of phosphorescent plates placed in the vessel at varying distances apart. With the plates 1 mm. apart the effect was very feeble, but increased directly as the distance and was large for a distance of 3 cms.
253These effects receive a general explanation on the views already put forward. When the radium is placed in the closed vessel, the emanation is given off at a constant rate and gradually diffuses throughout the enclosure. Since the time taken for diffusion of the emanation through tubes of ordinary size is small compared with the time required for the activity to be appreciably reduced, the emanation, and also the excited activity due to it, will be nearly equally distributed throughout the vessel.
The luminosity due to it should thus be equal at each end of the tube. Even with a capillary tube connecting the two bulbs, the gas continuously given off by the radium will always carry the emanation with it and cause a practically uniform distribution.
The gradual increase of the amount of emanation throughout the tube will be given by the equation
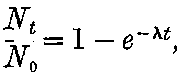
where Nt is the number of emanation particles present at the time t, N₀ the number present when radio-active equilibrium is reached, and λ is the radio-active constant of the emanation. The phosphorescent action, which is due partly to the radiations from the emanation and partly to the excited activity on the walls, should thus reach half the maximum value in four days and should practically reach its limit after three weeks’ interval.
The variation of luminosity with different distances between the screens is to be expected. The amount of excited activity deposited on the boundaries is proportional to the amount of emanation present. Since the emanation is equally distributed, the amount of excited activity deposited on the screens, due to the emanation between them, varies directly as the distance, provided the distance between the screens is small compared with their dimensions. Such a result would also follow if the phosphorescence were due to the radiation from the emanation itself, provided that the pressure of the gas was low enough to prevent absorption of the radiation from the emanation in the gas itself between the screens.
149. Emanating power. The compounds of thorium in the solid state vary very widely in the amount of emanation they emit under ordinary conditions. It is convenient to use the term emanating power to express the amount of emanation given off per second by one gram of the compound. Since, however, we have no means of determining absolutely the amount of emanation present, all measurements of emanating power are of necessity comparative. In most cases, it is convenient to take a given weight of a thorium compound, kept under conditions as nearly as possible constant, and to compare the amount of emanation of the compound to be examined with this standard.
In this way comparisons of the emanating power of thorium compounds have been made by Rutherford and Soddy[244], using an apparatus similar to that shown in Fig. 51 on page 240.
A known weight of the substance to be tested was spread on a shallow dish, placed in the glass tube C. A stream of dry dust-free air, kept constant during all the experiments, was passed over the compound and carried the emanation into the testing vessel. After ten minutes interval, the current due to the emanation in the testing vessel reached a constant value. The compound was then removed, and the standard comparison sample of equal weight substituted; the saturation current was observed when a steady state was again reached. The ratio of these two currents gives the ratio of the emanating power of the two samples.
It was found experimentally that, for the velocities of air current employed, the saturation current in the testing vessel was directly proportional to the weight of thorium, for weights up to 20 grams. This is explained by the supposition that the emanation is removed by the current of air from the mass of the compound, as fast as it is formed.
255By means of this relation the emanating power of compounds which are not of equal weight can be compared.
It was found that thorium compounds varied enormously in emanating power, although the percentage proportion of thorium present in the compound was not very different. For example, the emanating power of thorium hydroxide was generally 3 to 4 times greater than that of ordinary thoria, obtained from the manufacturer. Thorium nitrate, in the solid state, had only ¹⁄₂₀₀ of the emanating power of ordinary thoria, while preparations of the carbonate were found to vary widely among themselves in emanating power, which depended upon slight variations in the method of preparation.
150. Effect of conditions on emanating power. The emanating power of different compounds of thorium and radium is much affected by the alteration of chemical and physical conditions. In this respect the emanating power, which is a measure of the rate of escape of the emanation into the surrounding gas, must not be confused with the rate of decay of the activity of the emanations themselves, which has already been shown to be unaffected by external conditions.
Dorn (loc. cit.) first observed that the emanating power of thorium and radium compounds was much affected by moisture. In a fuller investigation of this point by Rutherford and Soddy, it was found that the emanating power of thoria is from two to three times greater in a moist than in a dry gas. Continued desiccation of the thoria in a glass tube, containing phosphorus pentoxide, did not reduce the emanating power much below that observed in ordinary dry air. In the same way radium chloride in the solid state gives off very little emanation when in a dry gas, but the amount is much increased in a moist gas.
The rate of escape of emanation is much increased by solution of the compound. For example, thorium nitrate, which has an emanating power of only ¹⁄₂₀₀ that of thoria in the solid state, has in solution an emanating power of 3 to 4 times that of thoria. P. Curie and Debierne observed that the emanating power of radium was also much increased by solution.
Temperature has a very marked effect on the emanating power. 256The writer[245] showed that the emanating power of ordinary thoria was increased three to four times by heating the substance to a dull red heat in a platinum tube. If the temperature was kept constant the emanation continued to escape at the increased rate, but returned to its original value on cooling. If, however, the compound was heated to a white heat, the emanating power was greatly reduced, and it returned on cooling to about 10% of the original value. Such a compound is said to be de-emanated. The emanating power of radium compounds varies in a still more striking manner with rise of temperature. The rate of escape of the emanation is momentarily increased even 10,000 times by heating to a dull red heat. This effect does not continue, for the large escape of the emanation by heating is in reality due to the release of the emanation stored up in the radium compound. Like thoria, when the compound has once been heated to a very high temperature, it loses its emanating power and does not regain it. It regains its power of emanating, however, after solution and re-separation.
A further examination of the effect of temperature was made by Rutherford and Soddy[246]. The emanating power of thoria decreases very rapidly with lowering of temperature, and at the temperature of solid carbonic acid it is only about 10% of its ordinary value. It rapidly returns to its original value when the cooling agent is removed.
Increase of temperature from 80° C. to a dull red heat of platinum thus increases the emanating power about 40 times, and the effects can be repeated again and again, with the same compound, provided the temperature is not raised to the temperature at which de-emanation begins. De-emanation sets in above a red heat, and the emanating power is then permanently diminished, but even long-continued heating at a white heat never entirely destroys the emanating power.
151. Regeneration of emanating power. An interesting question arises whether the de-emanation of thorium and radium is due to a removal or alteration of the substance which produces the 257emanation, or whether intense ignition merely changes the rate of escape of the emanation from the solid into the surrounding atmosphere.
It is evident that the physical properties of the thoria are much altered by intense ignition. The compound changes in colour from white to pink; it becomes denser and also far less readily soluble in acids. In order to test if the emanating power could be regenerated by a cyclic chemical process, the de-emanated thoria was dissolved, precipitated as hydroxide and again converted into oxide. At the same time a specimen of the ordinary oxide was subjected to an exactly parallel process. The emanating power of both these compounds was the same, and was from two to three times greater than that of ordinary thoria.
Thus de-emanation does not permanently destroy the power of thorium of giving out an emanation, but merely produces an alteration of the amount of the emanation which escapes from the compound.
152. Rate of production of the emanation. The emanating power of thorium compounds, then, is a very variable quantity, much affected by moisture, heat, and solution. Speaking generally, increased temperatures and solution greatly increase the emanating power of both thorium and radium.
The wide differences between the emanating powers of these substances in the solid state and in solution pointed to the conclusion that the differences were probably due to the rate of escape of the emanation into the surrounding gas, and not to a variation of the rate of reaction which gave rise to the emanation. It is obvious that a very slight retardation in the rate of escape of the thorium emanation from the compound into the gas, will, on account of the rapid decay of activity of the emanation, produce great changes in emanating power. The regeneration of the emanating power of de-emanated thoria and radium by solution and chemical treatment made it evident that the original power of thorium and radium of producing the emanation still persisted in an unaltered degree.
The question whether the emanation was produced at the same rate in emanating as in non-emanating compounds can be put to a 258sharp quantitative test. If the rate of production of emanation goes on at the same rate in the solid compound where very little escapes, as in the solution where probably all escapes, the emanation must be occluded in the compound, and consequently there must be a sudden release of this emanation on solution of the compound. On account of the very slow decay of the activity of the emanation of radium, the effects should be far more marked in that compound than in thorium.
From the point of view developed in section 133, the exponential law of decay of the emanation expresses the result that Nt the number of particles remaining unchanged at the time t is given by
where N₀ is the initial number of particles present. When a steady state is reached, the rate of production q₀ of fresh emanation particles is exactly balanced by the rate of change of the particles N₀ already present, i.e.
N₀ in this case represents the amount of emanation “occluded” in the compound. Substituting the value of λ found for the radium emanation in section 145,
The amount of emanation stored in a non-emanating radium compound should therefore be nearly 500,000 times the amount produced per second by the compound. This result was tested in the following way[247].
A weight of ·03 gr. of radium chloride of activity 1000 times that of uranium was placed in a Drechsel bottle and a sufficient amount of water drawn in to dissolve it. The released emanation was swept out by a current of air into a small gas holder and then into a testing cylinder. The initial saturation current was proportional to N₀. A rapid current of air was then passed through the radium solution for some time in order to remove any slight amount of emanation which had not been removed initially. The Drechsel bottle was 259closed air-tight, and allowed to stand undisturbed for a definite time t. The accumulated emanation was then swept out as before into the testing vessel. The new ionization current represents the value of Nt the amount of emanation formed in the compound during the interval t.
Assuming that there is no decay during the interval,
Making the small correction for the decay of activity during the interval,
We have previously shown that from the theory
The agreement between theory and experiment is thus as close as could be expected from the nature of the experiments. This experiment proves conclusively that the rate of production of emanation in the solid compound is the same as in the solution. In the former case it is occluded, in the latter it escapes as fast as it is produced.
It is remarkable how little emanation, compared with the amount stored up in the compound, escapes from solid radium chloride in a dry atmosphere. One experiment showed that the emanating power in the dry solid state was less than ½% of the emanating power of the solution. Since nearly 500,000 times as much emanation is stored up in the solid compound as is produced per second, this result showed that the amount of emanation which escaped per second was less than 10-8 of that occluded in the compound.
260If a solid radium chloride compound is kept in a moist atmosphere, the emanating power becomes comparable with the amount produced per second in the solution. In such a case, since the rate of escape is continuous, the amount occluded will be much less than the amount for the non-emanating material.
The phenomenon of occlusion of the radium emanation is probably not connected in any way with its radio-activity, although this property has here served to measure it. The occlusion of helium by minerals presents almost a complete analogy to the occlusion of the radium emanation. Part of the helium is given off by fergusonite, for example, when it is heated and all of it when the mineral is dissolved.
153. Similar results hold for thorium, but, on account of the rapid loss of activity of the emanation, the amount of emanation occluded in a non-emanating compound is very small compared with that observed for radium. If the production of the thorium emanation proceeds at the same rate under all conditions, the solution of a solid non-emanating compound should be accompanied by a rush of emanation greater than that subsequently produced. With the same notation as before we have for the thorium emanation,
This result was tested as follows: a quantity of finely powdered thorium nitrate, of emanating power ¹⁄₂₀₀ of ordinary thoria, was dropped into a Drechsel bottle containing hot water and the emanation rapidly swept out into the testing vessel by a current of air. The ionization current rose quickly to a maximum, but soon fell again to a steady value; showing that the amount of emanation released when the nitrate dissolves, is greater than the subsequent amount produced from the solution.
The rapid loss of the activity of the thorium emanation makes a quantitative comparison like that for radium very difficult. By slightly altering the conditions of the experiment, however, a definite proof was obtained that the rate of production of emanation is the same in the solid compound as in the solution. After dropping in the nitrate, a rapid air stream was blown through the 261solution for 25 seconds into the testing vessel. The air stream was stopped and the ionization current immediately measured. The solution was then allowed to stand undisturbed for 10 minutes. In that time the accumulation of the emanation again attained a practical maximum and again represented a steady state. The stream of air was blown through, as before, for 25 seconds, stopped and the current again measured. In both cases, the electrometer recorded a movement of 14·6 divisions per second. By blowing the same stream of air continuously through the solution the final current corresponded to 7·9 divisions per second or about one-half of that observed after the first rush.
Thus the rate of production of emanation is the same in the solid nitrate as in the solution, although the emanating power, i.e. the rate of escape of the emanation, is over 600 times greater in the solution than in the solid.
It seems probable that the rate of production of emanation by thorium, like the rate of production of Ur X and Th X, is independent of conditions. The changes of emanating power of the various compounds by moisture, heat, and solution must therefore be ascribed solely to an alteration in the rate of escape of the emanation into the surrounding gas and not to an alteration in the rate of its production in the compound.
On this view, it is easy to see that slight changes in the mode of preparation of a thorium compound may produce large changes in emanating power. Such effects have been often observed, and must be ascribed to slight physical changes in the precipitate. The fact that the rate of production of the emanation is independent of the physical or chemical conditions of the thorium, in which it is produced, is thus in harmony with what had previously been observed for the radio-active products Ur X and Th X.
154. Some experiments of Rutherford and Soddy[248] will now be considered, which show that the thorium emanation is produced, not directly by the thorium itself, but by the active product Th X.
262When the Th X, by precipitation with ammonia, is removed from a quantity of thorium nitrate, the precipitated thorium hydroxide does not at first possess appreciable emanating power. This loss of emanating power is not due, as in the case of the de-emanated oxide, to a retardation in the rate of escape of the emanation produced; for the hydroxide, when dissolved in acid, still gives off no emanation. On the other hand, the solution, containing the Th X, possesses emanating power to a marked degree. When the precipitated hydroxide and the Th X is left for some time, it is found that the Th X decreases in emanating power, while the hydroxide gradually regains its emanating power. After about a month’s interval, the emanating power of the hydroxide has nearly reached a maximum, while the emanating power of the Th X has almost disappeared.
The curves of decay and recovery of emanating power with time are found to be exactly the same as the curves of decay and recovery of activity of Th X and the precipitated hydroxide respectively, shown in Fig. 47. The emanating power of Th X, as well as its activity, falls to half value in four days, while the hydroxide regains half its final emanating power as well as half its lost activity in the same interval.
It follows from these results that the emanating power of Th X is directly proportional to its activity, i.e. that the rate of production of emanating particles is always proportional to the number of α particles, projected from the Th X per second. The radiation from Th X thus accompanies the change of the Th X into the emanation. Since the emanation has chemical properties distinct from those of the Th X, and also a distinctive rate of decay, it cannot be regarded as a vapour of Th X, but it is a distinct chemical substance, produced by the changes occurring in Th X. On the view advanced in section 136, the atom of the emanation consists of the part of the atom of Th X left behind after the expulsion of one or more α particles. The atoms of the emanation are unstable, and in turn expel α particles. This projection of α particles constitutes the radiation from the emanation, which serves as a measure of the amount of emanation present. Since the activity of the emanation falls to half value in one minute while that of Th X falls to half value in four days, the emanation 263consists of atoms which disintegrate at intervals nearly 6000 times shorter than those of the atoms of Th X.
155. No intermediate stage—Radium X—between radium and its emanation, corresponding to the Th X for thorium, has so far been observed. The emanation from radium is probably produced directly from that element. In this respect, the radium emanation holds the same position in regard to radium as Th X does to thorium, and its production from radium can be explained on exactly similar lines. It will be shown later in chapter X, that the emanation of actinium, like that of thorium, does not arise directly from the parent element but from an intermediate product actinium X, which is very analogous in physical and chemical properties to Th X.
156. Special methods are necessary to examine the nature of the radiation from the emanations, for the radiations arise from the volume of the gas in which the emanations are distributed. Some experiments to examine the radiations from the thorium emanation were made by the writer in the following way.
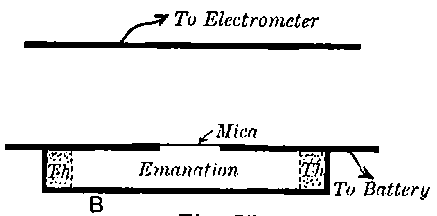
Fig. 55.
A highly emanating thorium compound wrapped in paper was placed inside a lead box B about 1 cm. deep, shown in Fig. 55. An opening was cut in the top of the box, over which a very thin sheet of mica was waxed. The emanation rapidly diffused through the paper into the vessel, and after ten minutes reached a state of radio-active equilibrium. The penetrating power of the radiation from the emanation which passed through the thin mica window was examined by the electrical method in the usual way by adding 264screens of thin aluminium foil. The results are expressed in the following table:
| Layers of foil | Current |
| 0 | 100 |
| 1 | 59 |
| 2 | 30 |
| 3 | 10 |
| 4 | 3·2 |
The greater proportion of the conductivity is thus due to α rays, as in the case of the radio-active elements. The amount of absorption of these α rays by aluminium foil is about the same as that of the rays from the active bodies. No direct comparison can be made, for the α rays from the emanation show the characteristic property of increased rate of absorption with thickness of matter traversed. Before testing, the rays have been largely absorbed by the mica window, and the penetrating power has consequently decreased.
No alteration in the radiation from the emanation was observed on placing an insulated wire inside the emanation vessel, and charging it to a high positive or negative potential. When a stream of air through the vessel carried away the emanation as fast as it was produced, the intensity of the radiation fell to a small fraction of its former value.
No evidence of any β rays in the radiations was found in these experiments, although a very small effect would have been detected. After standing some hours, however, β rays began to appear. These were due to the excited activity deposited on the walls of the vessel from the emanation, and not directly to the emanation itself.
The radium emanation, like that of thorium, only gives rise to α rays. This was tested in the following way[249]:
A large amount of emanation was introduced into a cylinder made of sheet copper ·005 cm. thick, which absorbed all the α rays but allowed the β and γ rays, if present, to pass through with but little loss. The external radiation from the cylinder 265was determined at intervals, commencing about two minutes after the introduction of the emanation. The amount observed at first was extremely small, but increased rapidly and practically reached a maximum in three or four hours. Thus the radium emanation only gives out α rays, the β rays appearing as the excited activity is produced on the walls of the vessel. On sweeping out the emanation by a current of air, there was no immediately appreciable decrease of the radiation. This is another proof that the emanation does not emit any β rays. In a similar way it can be shown that the emanation does not give out γ rays; these rays always make their appearance at the same time as the β rays.
The method of examination of the radiations from the emanations has been given in some detail, as the results are of considerable importance in the discussion, which will be given later in chapters X and XI, of the connection between the changes occurring in radio-active products and the radiations they emit. There is no doubt that the emanations, apart from the excited activity to which they give rise, only give out α rays, consisting most probably of positively charged bodies projected with great velocity.
157. It has already been mentioned that the conductivity due to the thorium emanation is proportional to the pressure of the gas, pointing to the conclusion that the rate of production of the emanation is independent of the pressure, as well as of the nature of the surrounding gas. This result was directly confirmed with the apparatus of Fig. 55. When the pressure of the gas under the vessel was slowly reduced, the radiation, tested outside the window, increased to a limit, and then remained constant over a wide range of pressure. This increase, which was far more marked in air than in hydrogen, is due to the fact that the α rays from the emanation were partially absorbed in the gas inside the vessel when at atmospheric pressure. At pressures of the order of 1 millimetre of mercury the external radiation decreased, but experiment showed that this must be ascribed to a removal of the emanation by the pump, and not to a change in the rate of 266production. The thorium compounds very readily absorb water-vapour, which is slowly given off at low pressures, and in consequence some of the emanation is carried out of the vessel with the water-vapour.
Curie and Debierne[250] found that both the amount of excited activity produced in a closed vessel containing active samples of radium, and also the time taken to reach a maximum value, were independent of the pressure and nature of the gas. This was true in the case of a solution down to the pressure of the saturated vapour, and in the case of solid salts to very low pressures. When the pump was kept going at pressures of the order of ·001 mm. of mercury, the amount of excited activity was much diminished. This was probably not due to any alteration of the rate of escape of the emanation, but to the removal of the emanation by the action of the pump as fast as it was formed.
Since the amount of excited activity, when in a state of radio-active equilibrium, is a measure of the amount of emanation producing it, these results show that the amount of emanation present when the rate of production balances the rate of decay is independent of the pressure and nature of the gas. It was also found that the time taken to reach the point of radio-active equilibrium was independent of the size of the vessel or the amount of active matter present. This proves that the state of equilibrium cannot in any way be ascribed to the possession by the emanation of any appreciable vapour pressure; for if such were the case, the time taken to reach the equilibrium value should depend on the size of the vessel and the amount of active matter present. The results are, however, in agreement with the view that the emanation is present in minute quantity in the tube, and that the equilibrium is governed purely by the radio-active constant λ, the constant of decay of activity of the emanation. This has been seen to be the same under all conditions of concentration, pressure and temperature, and, provided the rate of supply of the emanation from the active compound is not changed, the time-rate of increase of activity to the equilibrium value will always be the same, whatever the size of the vessel or the nature and pressure of the surrounding gas.
158. We shall now consider some experiments on the physical and chemical properties of the emanations themselves, without reference to the material producing them, in order to see if they possess any properties which connect them with any known kind of matter.
It was soon observed that the thorium emanation passed unchanged through acid solutions, and later the same result was shown to hold true in the case of both emanations for every reagent that was tried. Preliminary observations[251] showed that the thorium emanation, obtained in the usual way by passing air over thoria, passed unchanged in amount through a platinum tube heated electrically to the highest temperature obtainable. The tube was then filled with platinum-black, and the emanation passed through it in the cold, and with gradually increasing temperatures, until the limit was reached. In another experiment, the emanation was passed through a layer of red-hot lead-chromate in a glass tube. The current of air was replaced by a current of hydrogen, and the emanation was sent through red-hot magnesium-powder and red-hot palladium-black, and, by using a current of carbon dioxide, through red-hot zinc-dust. In every case the emanation passed through without sensible change in the amount. If anything, a slight increase occurred, owing to the time taken for the gas-current to pass through the tubes when hot being slightly less than when cold, the decay en route being consequently less. The only known gases capable of passing in unchanged amount through all the reagents employed are the recently discovered members of the argon family.
But another possible interpretation might be put upon the results. If the emanation were the manifestation of a type of excited radio-activity on the surrounding atmosphere, then, since from the nature of the experiments it was necessary to employ in each case as the atmosphere, a gas not acted on by the reagent employed, the result obtained might be expected. Red-hot magnesium would not retain an emanation consisting of radio-active hydrogen, nor red-hot zinc-dust an emanation consisting of radio-active 268carbon dioxide. The incorrectness of this explanation was shown in the following way. Carbon dioxide was passed over thoria, then through a T-tube, where a current of air met and mixed with it, both passing on to the testing-cylinder. But between this and the T-tube a large soda-lime tube was introduced, and the current of gas was thus freed from its admixed carbon dioxide, before being tested in the cylinder for the emanation. The amount of emanation found was quite unchanged, whether carbon dioxide was sent over thoria in the manner described, or whether, keeping the other arrangements as before, an equally rapid current of air was substituted for it. The theory that the emanation is an effect of the excited activity on the surrounding medium is thus excluded.
Experiments of a similar kind on the radium emanation were made later. A steady stream of gas was passed through a radium chloride solution and then through the reagent to be employed, into a testing-vessel of small volume, so that any change in the amount of emanation passing through could readily be detected. The radium emanation, like that of thorium, passed unchanged in amount through every reagent used.
In later experiments by Sir William Ramsay and Mr Soddy[252], the emanation from radium was exposed to still more drastic treatment. The emanation in a glass tube was sparked for several hours with oxygen over alkali. The oxygen was then removed by ignited phosphorus and no visible residue was left. When, however, another gas was introduced, mixed with the minute amount of emanation in the tube and withdrawn, the activity of emanation was found to be unaltered. In another experiment, the emanation was introduced into a magnesium lime tube, which was heated for three hours at a red heat. The emanation was then removed and tested, but no diminution in its discharging power was observed.
The emanations of thorium and radium thus withstand chemical treatment in a manner hitherto unobserved except in gases of the argon family.
159. Ramsay and Soddy (loc. cit.) record an interesting experiment to illustrate the gaseous nature of the emanation. 269A large amount of the radium emanation was collected in a small glass tube. This tube phosphoresced brightly under the influence of the rays from the emanation. The passage of the emanation from point to point was observed in a darkened room by the luminosity excited in the glass. On opening the stop-cock connecting with the Töpler pump, the slow flow through the capillary tube was noticed, the rapid passage along the wider tubes, the delay in passing through a plug of phosphorous pentoxide, and the rapid expansion into the reservoir of the pump. When compressed, the luminosity of the emanation increased, and became very bright as the small bubble containing the emanation was expelled through the fine capillary tube.
160. It has been shown that the emanations of thorium and radium behave like radio-active gases, distributed in minute amount in the air or other gas in which they are tested. With the small quantities of active material so far investigated, the emanations have not yet been collected in sufficient amount to determine their density. Although the molecular weight of the emanations cannot yet be obtained by direct chemical methods, an indirect estimate of it can be made by determining the rate of their inter-diffusion into air or other gases. The coefficients of inter-diffusion of various gases have long been known, and the results show that the coefficient of diffusion of one gas into another is, for the simpler gases, approximately inversely proportional to the square root of the product of their molecular weights. If, therefore, the coefficient of diffusion of the emanation into air is found to have a value, lying between that of two known gases A and B, it is probable that the molecular weight of the emanation lies between that of A and B.
Although the volume of the emanation given off from radium is very small, the electrical conductivity produced by the emanation in the gas, with which it is mixed, is often very large, and offers a ready means of measuring the emanation present.
Some experiments have been made by Miss Brooks and the writer[253] to determine the rate of the diffusion of the radium emanation 270into air, by a method similar to that employed by Loschmidt[254] in 1871, in his investigations of the coefficient of inter-diffusion of gases.
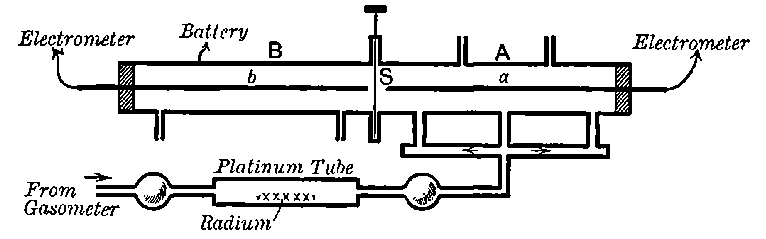
Fig. 56.
Fig. 56 shows the general arrangement. A long brass cylinder AB, of length 73 cms., and diameter 6 cms., was divided into two equal parts by a moveable metal slide S. The ends of the cylinder were closed with ebonite stoppers. Two insulated brass rods, a and b, each half the length of the tube, passed through the ebonite stoppers and were supported centrally in the tube. The cylinder was insulated and connected with one pole of a battery of 300 volts, the other pole of which was earthed. The central rods could be connected with a sensitive quadrant electrometer. The cylinder was covered with a thick layer of felt, and placed inside a metal box filled with cotton wool in order to keep temperature conditions as steady as possible.
In order to convey a sufficient quantity of emanation into the half-cylinder A, it was necessary to heat the radium slightly. The slide S was closed and the side tubes opened. A slow current of dry air from a gasometer was passed through a platinum tube, in which a small quantity of radium compound was placed. The emanation was carried with the air into the cylinder A. When a sufficient quantity had been introduced, the stream of air was stopped. The side tubes were closed by fine capillary tubes. These prevented any appreciable loss of gas due to the diffusion, but served to keep the pressure of the gas inside A at the pressure of the outside air. The three entrance tubes into the cylinder, shown in the figure, were for the purpose of initially mixing the emanation and gas as uniformly as possible.
271After standing several hours to make temperature conditions steady, the slide was opened, and the emanation began to diffuse into the tube B. The current through the tubes A and B was measured at regular intervals by an electrometer, with a suitable capacity in parallel. Initially there is no current in B, but after the opening of the slide, the amount in A decreased and the amount in B steadily increased. After several hours the amount in each half is nearly the same, showing that the emanation is nearly uniformly diffused throughout the cylinder.
It can readily be shown[255] that if
then
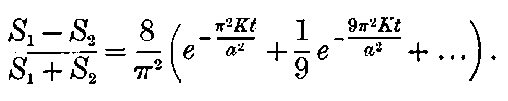
Now the values of S1 and S2 are proportional to the saturation ionization currents due to the emanations in the two halves of the cylinder. From this equation K can be determined, if the relative values of S1 and S2 are observed after diffusion has been in progress for a definite interval t.
The determination of S1 and S2 is complicated by the excited activity produced on the walls of the vessel. The ionization due to this must be subtracted from the total ionization observed in each half of the cylinder, for the excited activity is produced from the material composing the emanation, and is removed to the electrodes in an electric field. The ratio of the current due to excited activity to the current due to the emanation depends on the time of exposure to the emanation, and is only proportional to it for exposures of several hours.
The method generally adopted in the experiments was to open the slide for a definite interval, ranging in the experiments from 15 to 120 minutes. The slide was then closed and the currents in each half determined at once. The central rods, which had 272been kept negatively charged during the experiments, had most of the excited activity concentrated on their surfaces. These were removed, new rods substituted and the current immediately determined. The ratio of the currents in the half cylinders under these conditions was proportional to S1 and S2, the amounts of emanation present in the two halves of the cylinder.
The values of K, deduced from different values of t, were found to be in good agreement. In the earlier experiments the values of K were found to vary between ·08 and ·12. In some later experiments, where great care was taken to ensure that temperature conditions were very constant, the values of K were found to vary between ·07 and ·09. The lower value ·07 is most likely nearer the true value, as temperature disturbances tend to give too large a value of K. No certain differences were observed in the value of K whether the air was dry or damp, or whether an electric field was acting or not.
161. Some experiments on the rate of diffusion of the radium emanation into air were made at a later date by P. Curie and Danne[256]. If the emanation is contained in a closed reservoir, it has been shown that its activity, which is a measure of the amount of emanation present, decreases according to an exponential law with the time. If the reservoir is put in communication with the outside air through a capillary tube, the emanation slowly diffuses out, and the amount of emanation in the reservoir is found to decrease according to the same law as before, but at a faster rate. Using tubes of different lengths and diameters, the rate of diffusion was found to obey the same laws as a gas. The value of K was found to be 0·100. This is a slightly greater value of K than the lowest value 0·07 found by Rutherford and Miss Brooks. No mention is made by Curie and Danne of having taken any special precautions against temperature disturbances, and this may account for the higher value of K obtained by them.
They also found that the emanation, like a gas, always divided itself between two reservoirs, put in connection with one another, in the proportion of their volumes. In one experiment one reservoir was kept at a temperature of 10° C. and the other at 350° C. 273The emanation divided itself between the two reservoirs in the same proportion as would a gas under the same conditions.
162. For the purpose of comparison, a few of the coefficients of inter-diffusion of gases, compiled from Landolt and Bernstein’s tables, are given below.
| Gas or vapour | Coefficient of diffusion into air | Molecular weight |
|---|---|---|
| Water vapour | 0·198 | 18 |
| Carbonic acid gas | 0·142 | 44 |
| Alcohol vapour | 0·101 | 46 |
| Ether vapour | 0·077 | 74 |
| Radium emanation | 0·07 | ? |
The tables, although not very satisfactory for the purpose of comparison, show that the coefficient of inter-diffusion follows the inverse order of the molecular weights. The value of K for the radium emanation is slightly less than for ether vapour, of which the molecular weight is 74. We may thus conclude that the emanation is of greater molecular weight than 74. It seems likely that the emanation has a molecular weight somewhere in the neighbourhood of 100, and is probably greater than this, for the vapours of ether and alcohol have higher diffusion coefficients compared with carbonic acid than the theory would lead us to anticipate. Comparing the diffusion coefficients of the emanation and carbonic acid into air, the value of the molecular weight of the emanation should be about 176 if the result observed for the simple gases, viz. that the coefficient of diffusion is inversely proportional to the square root of the molecular weights, holds true in the present case. Bumstead and Wheeler[257] compared the rates of diffusion of the radium emanation and of carbon dioxide through a porous plate, and concluded that the molecular weight of the emanation was about 180. On the disintegration theory, the atom of the emanation is derived from the radium atom by the expulsion of one α particle. Thus, it is to be expected that its molecular weight would be over 200.
It is of interest to compare the value of K = ·07 with the value of K determined by Townsend (section 37) for the gaseous ions 274produced in air at ordinary pressure and temperature, by Röntgen rays or by the radiations from active substances. Townsend found that the value of K in dry air was ·028 for the positive ions and ·043 for the negative ions. The radium emanation thus diffuses more rapidly than the ions produced by its radiation in the gas, and behaves as if its mass were smaller than that of the ions produced in air, but considerably greater than that of the air molecules with which it is mixed.
It is not possible to regard the emanation as a temporarily modified condition of the gas originally in contact with the active body. Under such conditions a much larger value of K would be expected. The evidence derived from the experiments on diffusion strongly supports the view that the emanation is a gas of heavy molecular weight.
Makower[258] has recently attacked the question of the molecular weight of the radium emanation by another method. The rate of diffusion of the emanation through a porous plug of plaster-of-Paris was compared with that of the gases oxygen, carbon dioxide, and sulphur dioxide. It was found that Graham’s law, viz. that the coefficient of diffusion K is inversely proportional to the square root of its molecular weight M, was not strictly applicable. The value of K √M was not found to be constant for these gases, but decreased with increase of molecular weight of the gas. If, however, a curve was plotted with K √M as ordinate and K as abscissa, the points corresponding to the values of O, CO2 and SO2 were found to lie on a straight line. By linear extrapolation, the molecular weight of the emanation was estimated. The value obtained from experiments on three different porous plugs was 85·5, 97, and 99 respectively. This method indicates that the molecular weight of the radium emanation is about 100; but in all the experiments on diffusion, it must be remembered that the emanation, whose rate of inter-diffusion is being examined, exists in minute quantity mixed with the gas, and is compared with the rate of inter-diffusion of gases which are present in large quantity. For this reason, deductions of the molecular weight of the emanation may be subject to comparatively large errors, for which it is difficult to make correction.
163. On account of the rapid decay of the activity of the thorium emanation, it is not possible to determine the value of K its coefficient of diffusion into air by the methods employed for the radium emanation. The value of K has been determined by the writer in the following way. A plate C, Fig. 57, covered with thorium hydroxide, was placed horizontally near the base of a long vertical brass cylinder P. The emanation released from the thorium compound diffuses upwards in the cylinder.
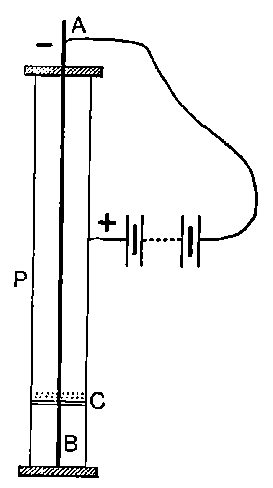
Fig. 57.
Let p be the partial pressure of the emanation at a distance x from the source C. This will be approximately uniform over the cross section of the cylinder. From the general principles of diffusion we get the equation
The emanation is continuously breaking up and expelling α particles. The emanation-residue gains a positive charge, and, in an electric field, is removed at once from the gas to the negative electrode.
Since the activity of the emanation at any time is always proportional to the number of particles which have not broken up, and since the activity decays with the time according to an exponential law,
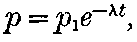
where p1 is the value of p when t = 0 and λ is the radio-active constant of the emanation.
Thus
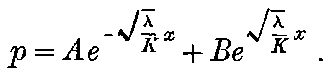
Since p = 0 when x = ∞. B = 0. If p = p₀ when x = 0, A = p₀.
Thus
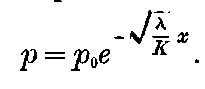
276It was not found convenient in the experiments to determine the activity of the emanation along the cylinder, but an equivalent method was used which depends upon measuring the distribution of “excited activity,” produced along a central rod AB, which is charged negatively.
It will be shown later (section 177) that the amount of excited activity at any point is always proportional to the amount of emanation at that point. The distribution of “excited activity” along the central rod from the plate C upwards thus gives the variation of p for the emanation along the tube.
In the experiments, the cylinder was filled with dry air at atmospheric pressure and was kept at a constant temperature. The central rod was charged negatively and exposed from one to two days in the presence of the emanation. The rod was then removed, and the distribution of the excited activity along it determined by the electric method. It was found that the amount of excited activity fell off with the distance x according to an exponential law, falling to half value in about 1·9 cms. This is in agreement with the above theory.
Since the activity of the emanation falls to half value in 1 minute, λ = ·0115. The value K = ·09 was deduced from the average of a number of experiments. This is a slightly greater value than K = ·07, obtained for the radium emanation, but the results show that the two emanations do not differ much from one another in molecular weight.
Makower (loc. cit.) compared the rates of diffusion of the thorium and radium emanation through a porous plate, and concluded that the two emanations were of about the same molecular weight, thus confirming the results obtained by the above method.
164. Experiments have been made by Wallstabe[259] on the coefficient of diffusion of the radium emanation into various liquids. The radium emanation was allowed to diffuse into a closed reservoir, containing a cylinder of the liquid under observation. The cylinder 277was provided with a tube and a stop-cock extending beyond the closed vessel, so that different layers of the liquid could be removed. The liquid was then placed in a closed testing vessel, where the ionization current due to the escape of the emanation from the liquid was observed to rise to a maximum after several hours, and then to decay. This maximum value of the current was taken as a measure of the amount of emanation absorbed in the liquid.
The coefficient of diffusion K of the emanation into the liquid can be obtained from the same equation used to determine the diffusion of the thorium emanation into air,
where λ is the constant of decay of activity of the radium emanation and x the depth of the layer of water from the surface.
Putting
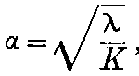
it was found that
The value of λ expressed in terms of a day as the unit of time is about ·17.
Thus the value of K for the diffusion of the radium emanation into water = ·066 cm.2 / day.
The value of K found by Stefan[260] for the diffusion of carbon dioxide into water was 1·36 cm.2/day. These results are thus in harmony with the conclusion drawn from the diffusion of the radium emanation into air, and show that the radium emanation behaves as a gas of high molecular weight.
165. Condensation of the emanations. During an investigation of the effect of physical and chemical agencies on the thorium emanation, Rutherford and Soddy[261] found that the 278emanation passed unchanged in amount through a white-hot platinum tube and through a tube cooled to the temperature of solid carbon dioxide. In later experiments the effects of still lower temperatures were examined, and it was then found that at the temperature of liquid air both emanations were condensed[262].
If either emanation is conveyed by a slow stream of hydrogen, oxygen, or air through a metal spiral immersed in liquid air, and placed in connection with a testing vessel as in Fig. 51, no trace of emanation escapes in the issuing gas. When the liquid air is removed and the spiral plunged into cotton-wool, several minutes elapse before any deflection of the electrometer needle is observed, and then the condensed emanation volatilizes rapidly, and the movement of the electrometer needle is very sudden, especially in the case of radium. With a fairly large amount of radium emanation, under the conditions mentioned, a very few seconds elapse after the first sign of movement before the electrometer needle indicates a deflection of several hundred divisions per second. It is not necessary in either case that the emanating compound should be retained in the gas stream. After the emanation is condensed in the spiral, the thorium or radium compound may be removed and the gas stream sent directly into the spiral. But in the case of thorium, under these conditions, the effects observed are naturally small owing to the rapid loss of the activity of the emanation with time, which proceeds at the same rate at the temperature of liquid air as at ordinary temperatures.
If a large amount of radium emanation is condensed in a glass U tube, the progress of the condensation can be followed by the eye, by means of the phosphorescence which the radiations excite in the glass. If the ends of the tube are sealed and the temperature allowed to rise, the glow diffuses uniformly throughout the tube, and can be concentrated at any point to some extent by local cooling of the tube with liquid air.
166. Experimental arrangements. A simple experimental arrangement to illustrate the condensation and volatilization of the emanation and some of its characteristic properties is shown in 279Fig. 58. The emanation obtained from a few milligrams of radium bromide by solution or heating is condensed in the glass U tube T immersed in liquid air. This U tube is then put into connection with a larger glass tube V, in the upper part of which is placed a piece of zinc sulphide screen Z, and in the lower part of the tube a piece of the mineral willemite. The stop-cock A is closed and the U tube and the vessel V are partially exhausted by a pump through the stop-cock B. This lowering of the pressure causes a more rapid diffusion of the emanation when released. The emanation does not escape if the tube T is kept immersed in liquid air. The stop-cock B is then closed, and the liquid air removed. No luminosity of the screen or the willemite in the tube V is observed for several minutes, until the temperature of T rises above the point of volatilization of the emanation. The emanation is then rapidly carried into the vessel V, partly by expansion of the gas in the tube T with rising temperature, and partly by the process of diffusion. The screen Z and the willemite W are caused to phosphoresce brilliantly under the influence of the rays from the emanation surrounding them.
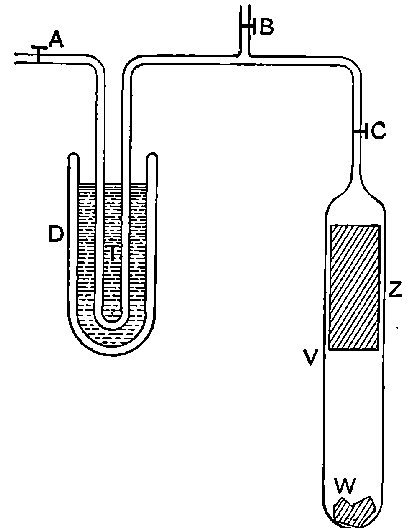
Fig. 58.
If the end of the vessel V is then plunged into liquid air, the emanation is again condensed in the lower end of the tube, and the willemite phosphoresces much more brightly than before. This is not due to an increase of the phosphorescence of willemite at the temperature of the liquid air, but to the effect of the rays from the emanation condensed around it. At the same time the luminosity of the zinc sulphide gradually diminishes, and practically disappears after several hours if the end of the tube is kept in the liquid air. If the tube is removed from the liquid air, the emanation again volatilizes and lights up the screen Z. The luminosity of the willemite returns to its original value after the lapse of several hours. This slow change of the luminosity of the zinc sulphide screen and of the willemite is due to the gradual 280decay of the “excited activity” produced by the emanation on the surface of all bodies exposed to its action (chapter VIII). The luminosity of the screen is thus due partly to the radiation from the emanation and partly to the excited radiation caused by it. As soon as the emanation is removed from the upper to the lower part of the tube, the “excited” radiation gradually diminishes in the upper and increases in the lower part of the tube.
The luminosity of the screen gradually diminishes with the time as the enclosed emanation loses its activity, but is still appreciable after an interval of several weeks.
An apparatus of a similar character to illustrate the condensation of the radium emanation has been described by P. Curie[263].
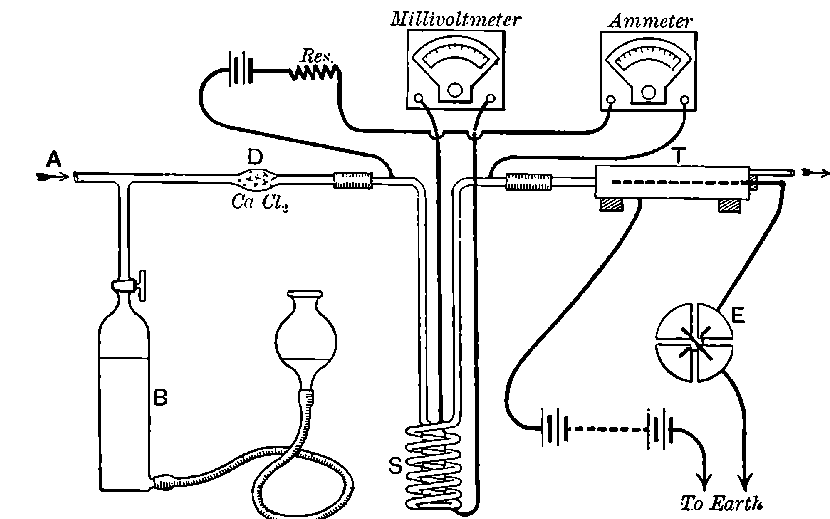
Fig. 59.
167. Determination of the temperature of condensation. A detailed investigation was made by Rutherford and Soddy (loc. cit.) of the temperatures at which condensation and volatilization commenced for the two emanations. The experimental arrangement of the first method is shown clearly in Fig. 59. A slow constant stream of gas, entering at A, was passed through a copper spiral S, over 3 metres in length, immersed in a bath of liquid ethylene. The copper spiral was made to act as its own thermometer by determining its electrical resistance. The 281resistance temperature curve was obtained by observation of the resistances at 0°, the boiling point of liquid ethylene -103·5°, the solidification point of ethylene -169° and in liquid air. The temperature of the liquid air was deduced from the tables given by Baly for the boiling point of liquid air for different percentages of oxygen. The resistance-temperature curve, for the particular spiral employed, was found to be nearly a straight line between 0° and -192°C., cutting the temperature axis if produced nearly at the absolute zero. The resistance of the spiral, deduced from readings on an accurately calibrated Weston millivoltmeter, with a constant current through the spiral, was thus very approximately proportional to the absolute temperature. The liquid ethylene was kept vigorously stirred by an electric motor, and was cooled to any desired temperature by surrounding the vessel with liquid air.
The general method employed for the radium emanation was to pass a suitable amount of emanation, mixed with the gas to be used, from the gas holder B into the spiral, cooled below the temperature of condensation. After the emanation was condensed in the spiral, a current of electrolytic hydrogen or oxygen was passed through the spiral. The temperature was allowed to rise gradually, and was noted at the instant when a deflection of the electrometer, due to the presence of emanation in the testing vessel T, was observed. The resistance, subject to a slight correction due to the time taken for the emanation to be carried into the testing vessel, gave the temperature at which some of the emanation commenced to volatilize. The ionization current in the testing vessel rose rapidly to a maximum value, showing that, for a small increase of temperature, the whole of the radium emanation was volatilized. The following table gives an illustration of the results obtained for a current of hydrogen of 1·38 cubic centimetres per second.
| Temperature | Divisions per second of the electrometer |
|---|---|
| -160° | 0 |
| -156° | 0 |
| -154°·3 | 1 |
| -153°·8 | 21 |
| -152°·5 | 24 |
282The following table shows the results obtained for different currents of hydrogen and oxygen.
| Current of Gas | T1 | T2 | |
|---|---|---|---|
| Hydrogen | ·25 c.c. per sec. | -151·3 | -150 |
| “ | ·32 „ „ | -153·7 | -151 |
| ” | ·92 „ „ | -152 | -151 |
| “ | 1·38 „ „ | -154 | -153 |
| ” | 2·3 „ „ | -162·5 | -162 |
| Oxygen | ·34 „ „ | -152·5 | -151·5 |
| “ | ·58 „ „ | -155 | -153 |
The temperature T1 in the above table gives the temperature of initial volatilization, T2 the temperature for which half of the condensed emanation had been released. For slow currents of hydrogen and oxygen, the values of T1 and T2 are in good agreement. For a stream of gas as rapid as 2·3 cubic centimetres per second the value of T1 is much lower. Such a result is to be expected; for, in too rapid a stream, the gas is not cooled to the temperature of the spiral, and, in consequence, the inside surface of the spiral is above the mean temperature, and some of the emanation escapes at a temperature apparently much lower. In the case of oxygen, this effect appears for a gas stream of 0·58 cubic centimetres per second.
In the experiments on the thorium emanation, on account of the rapid loss of activity, a slightly different method was necessary. The steady stream of gas was passed over the thorium compound, and the temperature was observed at the instant when an appreciable movement of the electrometer appeared. This gave the temperature at which a small fraction of the thorium emanation escaped condensation, and not the value T1 observed for the radium emanation, which gave the temperature for which a small fraction of the previously condensed emanation was volatilized.
The following table illustrates the results obtained.
| Current of Gas | Temperature | |
|---|---|---|
| Hydrogen | ·71 c.c. per sec. | -155° C. |
| “ | 1·38 „ „ | -159° C. |
| Oxygen | ·58 „ „ | -155° C. |
283On comparing these results with the values obtained for the radium emanation, it will be observed that with equal gas streams the temperatures are nearly the same.
A closer examination of the thorium emanation showed, however, that this apparent agreement was only accidental, and that there was, in reality, a very marked difference in the effect of temperature on the two emanations. It was found experimentally that the radium emanation was condensed very near the temperature at which volatilization commenced, and that the points of condensation and volatilization were defined fairly sharply.
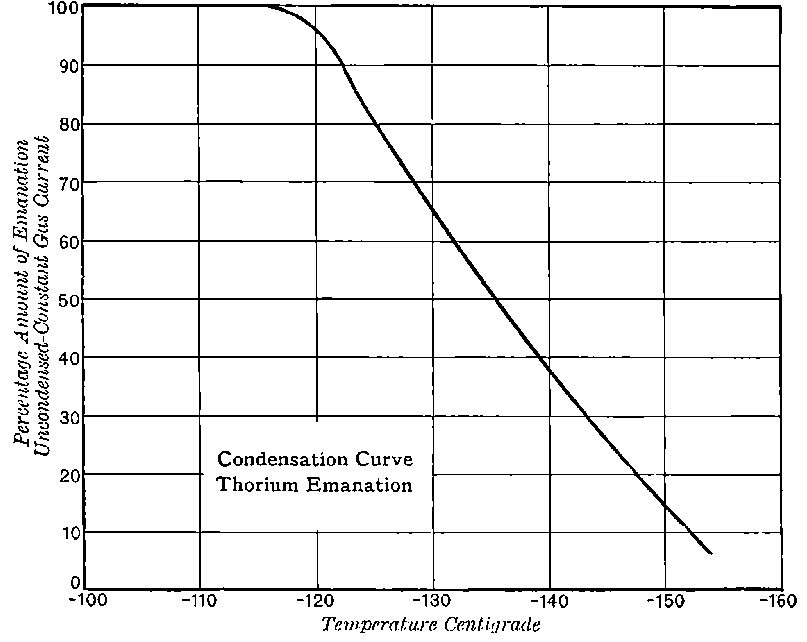
Fig. 60.
On the other hand, the thorium emanation required a range of over 30° C. after condensation had started in order to ensure complete condensation. Fig. 60 is an example of the results obtained with a steady gas stream of 1·38 c.c. per sec. of oxygen. The ordinates represent the percentage proportion of the emanation uncondensed at different temperatures. It will be observed that condensation commences about -120°, and that very little of the emanation escapes condensation at -155° C.
To investigate this difference of behaviour in the two emanations, a static method was employed, which allowed an examination 284of the two emanations to be made under comparable conditions. The emanation, mixed with a small amount of the gas to be used, was introduced into the cool spiral, which had been exhausted previously by means of a mercury pump. The amount of emanation remaining uncondensed after definite intervals was rapidly removed by means of the pump, and was carried with a constant auxiliary stream of gas into the testing vessel.
Tested in this way, it was found that the volatilization point of the radium emanation was very nearly the same as that obtained by the blowing method, viz. -150° C. With thorium, on the other hand, the condensation started at about -120° C., and, as in the blowing method, continued over a range of about 30° C. The proportion of the emanation condensed at any temperature was found to depend on a variety of conditions, although the point at which condensation commenced, viz. -120° C., was about the same in each case. It depended on the pressure and nature of the gas, on the concentration of the emanation, and on the time for which it was left in the spiral. For a given temperature a greater proportion of the emanation was condensed, the lower the pressure and the longer the time it was left in the spiral. Under the same conditions, the emanation was condensed more rapidly in hydrogen than in oxygen.
168. Thus there is no doubt that the thorium emanation begins to condense at a temperature higher than that at which the radium emanation condenses. The explanation of the peculiar behaviour of the thorium emanation is clear when the small number of emanation particles present in the gas are taken into consideration. It has been shown that both emanations give out only α rays. It is probable that the α particles from the two emanations are similar in character and produce about the same number of ions in their passage through the gas. The number of ions produced by each α particle before its energy is dissipated is probably about 70,000. (See section 252.)
Now, in the experiment, the electrometer readily measured a current of 10-3 electrostatic units. Taking the charge on an ion as 3·4 × 10-10 electrostatic units, this corresponds to a production in the testing vessel of about 3 × 106 ions per sec., which would be 285produced by about 40 expelled α particles per second. Each radiating particle cannot expel less than one α particle and may expel more, but it is likely that the number expelled by an atom of the thorium emanation is not greatly different from that expelled by an atom of the radium emanation.
In section 133 it has been shown that, according to the law of decay, λN particles change per second when N are present. Thus, to produce 40 α particles, λN cannot be greater than 40. Since for the thorium emanation λ is ¹⁄₈₇, it follows that N cannot be greater than 3500. The electrometer thus detected the presence of 3500 particles of the thorium emanation, and since in the static method the volume of the condensing spiral was about 15 c.c., this corresponded to a concentration of about 230 particles per c.c. An ordinary gas at atmospheric pressure and temperature probably contains about 3·6 × 1019 molecules per c.c. Thus the emanation would have been detected on the spiral if it had possessed a partial pressure of less than 10-17 of an atmosphere.
It is not surprising then that the condensation point of the thorium emanation is not sharply defined. It is rather a matter of remark that condensation should occur so readily with so sparse a distribution of emanation particles in the gas; for, in order that condensation may take place, it is probable that the particles must approach within one another’s sphere of influence.
Now in the case of the radium emanation, the rate of decay is about 5000 times slower than that of the thorium emanation, and consequently the actual number of particles that must be present to produce the same ionization per second in the two cases must be about 5000 times greater in the case of radium than in the case of thorium. This conclusion involves only the assumption that the same number of rays is produced by a particle of emanation in each case, and that the expelled particles produce in their passage through the gas the same number of ions. The number of particles present, in order to be detected by the electrometer, in this experiment, must therefore have been about 5000 × 3500, i.e. about 2 × 107. The difference of behaviour in the two cases is well explained by the view that, for equal electrical effects, the number of radium emanation particles must be far larger than the number of thorium 286emanation particles. The probability of the particles coming into each other’s sphere of influence will increase very rapidly as the concentration of the particles increases, and, in the case of the radium emanation, once the temperature of condensation is attained, all but a small proportion of the total number of particles present will condense in a very short time. In the case of the thorium emanation, however, the temperature might be far below that of condensation, and yet a considerable portion remain uncondensed for comparatively long intervals. On this view the experimental results obtained might reasonably be expected. A greater proportion of emanation condenses the longer the time allowed for condensation under the same conditions. The condensation occurs more rapidly in hydrogen than in oxygen, as the diffusion is greater in the former gas. For the same reason the condensation occurs faster the lower the pressure of the gas present. Finally, when the emanation is carried by a steady stream of gas, a smaller proportion condenses than in the other cases, because the concentration of emanation particles per unit volume of gas is less under these conditions.
It is possible that the condensation of the emanations may not occur in the gas itself but at the surface of the containing vessel. Accurate observations of the temperature of condensation have so far only been made in a copper spiral, but condensation certainly occurs in tubes of lead or glass at about the same temperature as in tubes of copper.
169. In experiments that were made by the static method with a very large quantity of radium emanation, a slight amount of escape of the condensed emanation was observed several degrees below the temperature at which most of the emanation was released. This is to be expected, since, under such conditions, the electrometer is able to detect a very minute proportion of the whole quantity of the emanation condensed.
Special experiments, with a large quantity of emanation, that were made with the spiral immersed in a bath of rapidly boiling nitric oxide, showed this effect very clearly. For example, the condensed emanation began to volatilize at -155° C. In 4 minutes the temperature had risen to -153·5°, and the amount volatilized 287was four times as great as at -155°. In the next 5-½ minutes the temperature had increased to -152·3° and practically the whole quantity, which was at least fifty times the amount at the temperature of -153·5°, had volatilized.
It thus seems probable that, if the temperature were kept steady at the point at which volatilization was first observed, and the released emanation removed at intervals, the whole of the emanation would in course of time be liberated at that temperature. Curie and Dewar and Ramsay have observed that the emanation condensed in a U tube, immersed in liquid air, slowly escapes if the pump is kept steadily working. These results point to the probability that the condensed emanation possesses a true vapour pressure, but great refinements in experimental methods would be necessary before such a conclusion could be definitely established.
The true temperature of condensation of the thorium emanation is probably about -120° C., and that of radium about -150° C. Thus there is no doubt that the two emanations are quite distinct from each other in this respect, and also with regard to their radio-activity, although they both possess the property of chemical inertness. These results on the temperatures of condensation do not allow us to make any comparison of the condensation points of the emanations with those of known gases, since the lowering of the condensation points of gases with diminution of pressure has not been studied at such extremely minute pressures.
170. It has been found[264] that the activity of the thorium emanation, when condensed in the spiral at the temperature of liquid air, decayed at the same rate as at ordinary temperatures. This is in accord with results of a similar kind obtained by P. Curie for the radium emanation (section 145), and shows that the value of the radio-active constant is unaffected by wide variations of temperature.
171. It has been shown in section 93 from experimental data that 1 gram of radium bromide at its minimum activity emits about 3·6 × 1010 α particles per second. Since the activity due to the emanation stored up in radium, when in a state of radio-active equilibrium, is about one quarter of the whole and about equal to the minimum activity, the number of α particles projected per second by the emanation from 1 gram of radium bromide is about 3·6 × 1010. It has been shown in section 152 that 463,000 times the amount of emanation produced per second is stored up in the radium. But, in a state of radio-active equilibrium, the number of emanation particles breaking up per second is equal to the number produced per second. Assuming that each emanation particle in breaking up expels one α particle, it follows that the number of emanation particles present in 1 gram of radium bromide in radio-active equilibrium is 463,000 × 3·6 × 1010, i.e. 1·7 × 1016. Taking the number of hydrogen molecules in 1 c.c. of gas at atmospheric pressure and temperature as 3·6 × 1019 (section 39), the volume of the emanation from 1 gram of radium bromide is 4·6 × 10-4 cubic centimetres at atmospheric pressure and temperature. Assuming the composition of radium bromide as RaBr2, the amount from 1 gram of radium in radio-active equilibrium is 0·82 cubic millimetres. Quite independently of any method of calculation it was early evident that the volume of the emanation was very small, for all the earlier attempts made to detect its presence by its volume were unsuccessful. It will be seen, however, that, when larger quantities of radium were available for experiment, the emanation has been collected in volume sufficiently large to measure.
In the case of thorium, the maximum quantity of emanation to be obtained from 1 gram of the solid is very minute, both on account of the small activity of thorium and of the rapid break up of the emanation after its production. Since the amount of emanation, stored in a non-emanating thorium compound, is only 87 times the rate of production, while in radium it is 463,000 times, and the rate of production of the emanation by radium is about 1 million 289times faster than by thorium, it follows that the amount of emanation to be obtained from 1 gram of thorium is not greater than 10-10 of the amount from an equal weight of radium, i.e. its volume is not greater than 10-13 c.c. at the ordinary pressure and temperature. Even with large quantities of thorium, the amount of emanation is too small ever to be detected by its volume.
172. Volume of the emanation from radium. The evidence already considered points very strongly to the conclusion that the emanation possesses all the properties of a chemically inert gas of high molecular weight.
Since the emanation continuously breaks up, and is transformed into a solid type of matter, which is deposited on the surface of bodies, the volume of the emanation, when separated from radium, should contract at the same rate as it loses its activity, i.e. it should decrease to half value in about four days. The amount of emanation to be obtained from a given quantity of radium is a maximum when the rate of production of new emanation balances its rate of change. This condition is practically attained when the emanation has been allowed to collect for an interval of one month. The probable volume of the emanation to be obtained from 1 gram of radium was early calculated on certain assumptions, and from data then available the writer[265] deduced that the volume of the emanation from 1 gram of radium lay between ·06 and ·6 cubic millimetre at atmospheric pressure and temperature, and was probably nearer the latter value. The volume to be expected on the latest data has been discussed in the preceding section and shown to be about ·82 cubic mm. The volume of the emanation is thus very small, but not too small to be detected if several centigrams of radium are available. This has been proved to be the case by Ramsay and Soddy[266] who, by very careful experiment, finally succeeded in isolating a small quantity of the emanation and in determining its volume. The experimental method employed by them will now be briefly described.
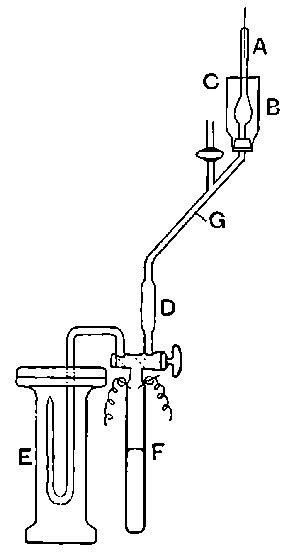
Fig. 61.
The emanation from 60 milligrams of radium bromide in solution was allowed to collect for 8 days and then drawn off through the inverted siphon E (Fig. 61) into the explosion burette F. This gas consisted for the most part of hydrogen and oxygen, produced by the action of the radiations on the water of the solution. After explosion, the excess of hydrogen mixed with emanation was left some time in contact with caustic soda, placed in the upper part of the burette, in order to remove all trace of carbon dioxide. In the meantime the upper part of the apparatus had been completely evacuated. The connection C to the pump was closed, and the hydrogen and emanation were allowed to enter the apparatus, passing over a phosphorous pentoxide tube D. The emanation was condensed in the lower part of the capillary tube A, by surrounding it with the tube B filled with liquid air. The process of condensation was rendered manifest by the brilliant luminosity of the lower part of the tube. The mercury from the burette was then allowed to run to G, and the apparatus again completely evacuated. The connection of the pump was again closed, the liquid air was removed and the volatilized emanation forced into the fine capillary tube A. Observations were then made, from day to day, of the volume of the emanation. The results are given in the table below.
| Time | Volume | Time | Volume |
|---|---|---|---|
| Start | 0·124 cub. mm. | 7 days | 0·0050 cub. mm. |
| 1 day | 0·027 „ | 9 „ | 0·0041 „ |
| 3 „ | 0·011 „ | 11 „ | 0·0020 „ |
| 4 „ | 0·0095 „ | 12 „ | 0·0011 „ |
| 6 „ | 0·0063 „ | 28 „ | 0·0004 „ |
The volume contracted with the time, and was very small after a month’s interval, but the minute bubble of the emanation still retained its luminosity to the last. The tube became deep purple in colour, which rendered readings difficult except with a strong light. There was a sudden decrease in the first day, 291which may have been due to the mercury sticking in the capillary tube.
The experiments were repeated with another capillary tube and the volume of gas observed at normal pressure was 0·0254 c. mm. The gas obtained was found to obey Boyle’s law within the limit of experimental error over a considerable range of pressure. But, unlike in the first experiment, the gas did not contract but expanded rapidly during the first few hours, and then more slowly, finally reaching a volume after 23 days of 0·262 c. mm. or about 10 times the initial volume. The measurements were complicated by the appearance of bubbles of gas in the top of the mercury column. The differences observed in these two experiments are difficult to account for. We shall see, later, that the emanation always produces helium, and, in the first experiment, the decrease of the volume to zero indicates that the helium was buried or absorbed in the walls of the tube. In the second case, probably owing to some difference in the glass of the capillary tube, the helium may have been released. This suggestion is confirmed by the observation that the volume of gas, after the experiment ended, gave a brilliant spectrum of helium.
We shall see later that there is considerable evidence that the α particles expelled from radio-active substances consist of helium atoms. Since the particles are projected with great velocity, they will first be buried in the walls of the tube, and then may gradually diffuse out into the gas again under conditions probably depending on the kind of glass employed. Since α particles are projected from the emanation and also from two of the rapidly changing products which arise from it, the volume of helium should, on this view, be three times the initial volume of the emanation. If the helium produced escaped from the walls of the tube into the gas, the apparent volume of the gas in the capillary should increase to three times the initial volume in a month’s interval, for during that time the emanation itself has been transformed into a solid type of matter deposited on the walls of the tube.
Ramsay and Soddy concluded from their experiments that the maximum volume of emanation to be obtained from 1 gram of radium was about 1 cubic millimetre at standard pressure 292and temperature, and that the emanation was produced from 1 gram of radium at the rate of 3 × 10-6 c. mm. per second. This amount is in very good agreement with the calculated value, and is a strong indication of the general correctness of the theory on which the calculations are based.
173. Spectrum of the emanation. After the separation of the emanation and the determination of its volume, Ramsay and Soddy made numerous attempts to obtain its spectrum. In some of the earlier experiments several bright lines were seen for a short time, but these lines were soon masked by the appearance of the hydrogen lines. In later experiments Ramsay and Collie[267] succeeded in obtaining a spectrum of the emanation, which persisted for a short time, during which a rapid determination of the wave-lengths was made. They state that the spectrum was very brilliant, consisting of very bright lines, the spaces between being perfectly dark. The spectrum bore a striking resemblance in general character to the spectrum of the gases of the argon family.
The spectrum soon faded, and the spectrum of hydrogen began to appear. The following table shows the wave-length of the lines observed in the spectrum. The degree of coincidence of the lines of known wave-lengths shows that the error is probably less than five Ångström units.
| Wave-length | Remarks |
|---|---|
| 6567 | Hydrogen C; true wave-length, 6563; observed each time. |
| 6307 | Observed only at first; evanescent. |
| 5975 | „ „ „ |
| 5955 | „ „ „ |
| 5805 | Observed each time; persistent. |
| 5790 | Mercury; true wave-length, 5790. |
| 5768 | „ „ 5769. |
| 5725 | Observed only at first; evanescent. |
| 5595 | Observed each time; persistent and strong. |
| 5465 | Mercury; true wave-length, 5461. |
| 5105 | Not observed at first; appeared after some seconds; persisted and was visible during the second examination. |
| 4985 | Observed each time; persistent and strong. |
| 4865 | Hydrogen F; true wave-length, 4861. |
| 4690 | Observed only at first. |
| 4650 | Not observed when the emanation was examined again. |
| 4630 | „ „ „ |
| 4360 | Mercury: true wave-length, 4359. |
293The experiments were repeated with a new supply of emanation, and some of the stronger lines were observed again, while some new lines made their appearance. Ramsay and Collie suggest that the strong line 5595 may be identical with a line which was observed by Pickering[268] in the spectrum of lightning, and was not identified with the spectrum of any known gas.
Until large quantities of radium are available for the experimenter it would appear difficult to make sure how many of these lines must be ascribed to the spectrum of the emanation or to measure the wave-lengths with accuracy.
The results are of great interest, as showing that the emanation has a definite and new spectrum of the same general character as the argon group of gases to which, as we have seen, it is chemically allied.
174. The investigations into the nature of the radio-active emanations have thus led to the following conclusions:—The radio-elements thorium, radium and actinium continuously produce from themselves radio-active emanations at a rate which is constant under all conditions. In some cases, the emanations continuously diffuse from the radio-active compounds into the surrounding gas; in other cases, the emanations are unable to escape from the material in which they are produced, but are occluded, and can only be released by solution or by the action of heat.
The emanations possess all the properties of radio-active gases. They diffuse through gases, liquids, and porous substances, and can be occluded in some solids. Under varying conditions of pressure, volume, and temperature, the emanations distribute themselves in the same way and according to the same laws as does a gas.
The emanations possess the important property of condensation under the influence of extreme cold, and by that means can be separated from the gases with which they are mixed. The radiation from the emanation is material in nature, and consists of a stream of positively charged particles projected with great velocity.
294The emanations possess the property of chemical inertness, and in this respect resemble the gases of the argon family. The emanations are produced in minute amount; but a sufficient quantity of the radium emanation has been obtained to determine its volume and its spectrum. With regard to their rates of diffusion, the emanations of both thorium and radium behave like gases of high molecular weight.
These emanations have been detected and their properties investigated by the property they possess of emitting radiations of a special character. These radiations consist entirely of α rays, i.e. particles, projected with great velocity, which carry a positive charge and have a mass about twice that of the hydrogen atom. The emanations do not possess the property of permanently radiating, but the intensity of the radiations diminishes according to an exponential law with the time, falling to half value, from actinium in 4 seconds, from thorium in one minute, and from radium in about four days. The law of decay of activity does not seem to be influenced by any physical or chemical agency.
The emanation particles gradually break up, each particle as it breaks up expelling a charged body. The emanation after it has radiated ceases to exist as such, but is transformed into a new kind of matter, which is deposited on the surface of bodies and gives rise to the phenomena of excited activity. This last property, and the connection of the emanation with it, are discussed in detail in the next chapter.
175. Excited radio-activity. One of the most interesting and remarkable properties of thorium, radium, and actinium, is their power of “exciting” or “inducing” temporary activity on all bodies in their neighbourhood. A substance which has been exposed for some time in the presence of radium or thorium behaves as if its surface were covered with an invisible deposit of intensely radio-active material. The “excited” body emits radiations capable of affecting a photographic plate and of ionizing a gas. Unlike the radio-elements themselves, however, the activity of the body does not remain constant after it has been removed from the influence of the exciting active material, but decays with the time. The activity lasts for several hours when due to radium and several days when due to thorium.
This property was first observed by M. and Mme. Curie[269] for radium, and independently by the writer[270] for thorium[271].
296If any solid body is placed inside a closed vessel containing an emanating compound of thorium or radium, its surface becomes radio-active. For thorium compounds the amount of excited activity on a body is in general greater the nearer it is to the active material. In the case of radium, however, provided the body has been exposed for several hours, the amount of excited activity is to a large extent independent of the position of the body in the vessel containing the active material. Bodies are made active whether exposed directly to the action of the radio-active substance or screened from the action of the direct rays. This has been clearly shown in some experiments of P. Curie. A small open vessel a (Fig. 62) containing a solution of radium is placed inside a larger closed vessel V.
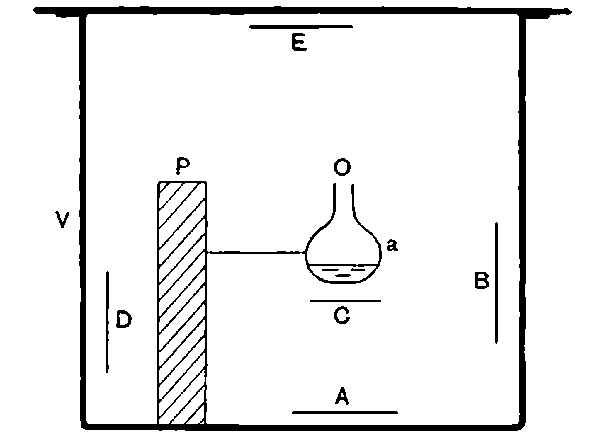
Fig. 62.
Plates A, B, C, D, E are placed in various positions in the enclosure. After exposure for a day, the plates after removal are found to be radio-active even in positions completely shielded from the action of the direct rays. For example, the plate D shielded from the direct radiation by the lead plate P is as active as the plate E, exposed to the direct radiation. The amount of activity produced in a given time on a plate of given area in a definite position is independent of the material of the plate. Plates of mica, copper, cardboard, ebonite, all show equal amounts of activity. The amount of activity depends on the area of the plate and on 297the amount of free space in its neighbourhood. Excited radio-activity is also produced in water if exposed to the action of an emanating compound.
176. Concentration of excited radio-activity on the negative electrode. When thorium or radium is placed in a closed vessel, the whole interior surface becomes strongly active. In a strong electric field, on the other hand, the writer found that the activity was confined entirely to the negative electrode. By suitable arrangements, the whole of the excited activity, which was previously distributed over the surface of the vessel, can be concentrated on a small negative electrode placed inside the vessel. An experimental arrangement for this purpose is shown in Fig. 63.
Fig. 63.
The metal vessel V containing a large amount of thoria is connected with the positive pole of a battery of about 300 volts. The wire AB to be made active is fastened to a stouter rod BC, passing through an ebonite cork inside a short cylinder D, fixed in the side of the vessel. This rod is connected with the negative pole of the battery. In this way the wire AB is the only conductor exposed in the field with a negative charge, and it is found that the whole of the excited activity is concentrated upon it.
In this way it is possible to make a short thin metal wire over 10,000 times as active per unit surface as the thoria from which the excited activity is derived. In the same way, the excited activity due to radium can be concentrated mainly on the negative 298electrode. In the case of thorium, if the central wire be charged positively, it shows no appreciable activity. With radium, however, a positively charged body becomes slightly active. In most cases, the amount of activity produced on the positive electrode is not more than 5% of the corresponding amount when the body is negatively charged. For both thorium and radium, the amount of excited activity on electrodes of the same size is independent of their material.
All metals are made active to equal extents for equal times of exposure. When no electric field is acting, the same amount of activity is produced on insulators like mica and glass as on conductors of equal dimensions.
177. Connection between the emanations and excited activity. An examination of the conditions under which excited activity is produced shows that there is a very close connection between the emanation and the excited activity. If a thorium compound is covered with several sheets of paper, which cut off the α rays but allow the emanation to pass through, excited activity is still produced in the space above it. If a thin sheet of mica is waxed down over the active material, thus preventing the escape of the emanation, no excited activity is produced outside it. Uranium and polonium which do not give off an emanation are not able to produce excited activity on bodies. Not only is the presence of the emanation necessary to cause excited activity, but the amount of excited activity is always proportional to the amount of emanation present. For example, de-emanated thoria produces very little excited activity compared with ordinary thoria. In all cases the amount of excited activity produced is proportional to the emanating power. When passing through an electric field the emanation loses its property of exciting activity at the same rate as the radiating power diminishes. This was shown by the following experiment.
A slow constant current of air from a gasometer, freed from dust by its passage through cotton-wool, passed through a rectangular wooden tube 70 cms. long. Four equal insulated metal plates A, B, C, D, were placed at regular intervals along the tube. The positive pole of a battery of 300 volts was connected with a metal 299plate placed in the bottom of the tube, while the negative pole was connected with the four plates. A mass of thoria was placed in the bottom of the tube under the plate A, and the current due to the emanation determined at each of the four plates. After passing a current of air of 0·2 cm. per second for 7 hours along the tube, the plates were removed and the amount of excited activity produced on them was tested by the electric method. The following results were obtained.
| Relative current due to emanation | Relative excited activity | |
|---|---|---|
| Plate A | 1 | 1 |
| „ B | ·55 | ·43 |
| „ C | ·18 | ·16 |
| „ D | ·072 | ·061 |
Within the errors of measurement, the amount of excited activity is thus proportional to the radiation from the emanation, i.e. to the amount of emanation present. The same considerations hold for the radium emanation. The emanation in this case, on account of the slow loss of its activity, can be stored mixed with air for long periods in a gasometer, and its effects tested quite independently of the active matter from which it is produced. The ionization current due to the excited activity produced by the emanation is always proportional to the current due to the emanation for the period of one month or more that its activity is large enough to be measured conveniently by an electrometer.
If, at any time during the interval, some of the emanation is removed and introduced into a new testing vessel, the ionization current will immediately commence to increase, rising in the course of four or five hours to about twice its original value. This increase of the current is due to the excited activity produced on the walls of the containing vessel. On blowing out the emanation, the excited activity is left behind, and at once begins to decay. Whatever its age, the emanation still possesses the property of causing excited activity, and in amount always proportional to its activity, i.e. to the amount of emanation present.
These results show that the power of exciting activity on 300inactive substances is a property of the radio-active emanations, and is proportional to the amount of emanation present.
The phenomenon of excited activity cannot be ascribed to a type of phosphorescence produced by the rays from the emanation on bodies; for it has been shown that the activity can be concentrated on the negative electrode in a strong electric field, even if the electrode is shielded from the direct radiation from the active substance which gives off the emanation. The amount of excited activity does not seem in any way connected with the ionization produced by the emanation in the gas with which it is mixed. For example, if a closed vessel is constructed with two large parallel insulated metal plates on the lower of which a layer of thoria is spread, the amount of the excited activity on the upper plate when charged negatively, is independent of the distance between the plates when that distance is varied from 1 millimetre to 2 centimetres. This experiment shows that the amount of excited activity depends only on the amount of emanation emitted from the thoria; for the ionization produced with a distance of 2 centimetres between the plates is about ten times as great as with a distance of 1 millimetre.
178. If a platinum wire be made active by exposure to the emanation of thoria, its activity can be removed by treating the wire with certain acids[272]. For example, the activity is not much altered by immersing the wire in hot or cold water or nitric acid, but more than 80% of it is removed by dilute or concentrated solutions of sulphuric or hydrochloric acid. The activity has not been destroyed by this treatment but is manifested in the solution. If the solution be evaporated, the activity remains behind on the dish.
These results show that the excited activity is due to a deposit on the surface of bodies of radio-active matter which has definite properties as regards solution in acids. This active matter is dissolved in some acids, but, when the solvent is evaporated, the active matter is left behind. This active matter is deposited on the surface of bodies, for it can be partly removed by rubbing the body with a cloth, and almost completely by scouring the plate 301with sand or emery paper. If a negatively charged wire is placed in the presence of a large quantity of radium emanation, it becomes intensely active. If the wire, after removal, is drawn across a screen of zinc sulphide, or willemite, a portion of the active matter is rubbed off, and a luminous trail is left behind on the screen. The amount of active matter deposited is extremely small, for no difference of weight has been detected in a platinum wire when made extremely active. On examining the wire under a microscope, no trace of foreign matter is observed. It follows from these results that the matter which causes excited activity is many thousand times more active, weight for weight, than radium itself.
It is convenient to have a definite name for this radio-active matter, for the term “excited activity” only refers to the radiation from the active matter and not to the matter itself. The term “active deposit” will be generally applied to this matter. The active deposit from the three substances thorium, radium, and actinium is, in each case, derived from its respective emanation, and possesses the same general property of concentration on the negative electrode in an electric field and of acting as a non-volatile type of matter which is deposited from the gas on to the surface of bodies. These active deposits, while all soluble in strong acids, are chemically distinct from each other.
The term “active deposit” can, however, only be used when the matter is spoken of as a whole; for it will be shown later that the matter, under ordinary conditions, is complex and contains several constituents which have distinctive physical and chemical properties and also a distinctive rate of change. According to the theory advanced in section 136, we may suppose that the emanation of thorium, radium, and actinium is unstable and breaks up with the expulsion of an α particle. The residue of the atom of the emanation diffuses to the sides of the vessel or is removed to the negative electrode in an electric field. This active deposit is in turn unstable and breaks up in several successive stages.
The “excited activity” proper is the radiation set up by the active deposit in consequence of the changes occurring in it. On this view, the emanation is the parent of the active deposit in the same way that Th X is the parent of the emanation. The 302proportionality which always exists between the activity of the emanation and the excited activity to which it gives rise, is at once explained, if one substance be the parent of the other.
179. Decay of the excited activity produced by thorium. The excited activity produced in a body after a long exposure to the emanations of thorium, decays in an exponential law with the time, falling to half value in about 11 hours. The following table shows the rate of decay of the excited activity produced on a brass rod.
| Time in hours | Current |
|---|---|
| 0 | 100 |
| 7·9 | 64 |
| 11·8 | 47·4 |
| 23·4 | 19·6 |
| 29·2 | 13·8 |
| 32·6 | 10·3 |
| 49·2 | 3·7 |
| 62·1 | 1·86 |
| 71·4 | 0·86 |
The results are shown graphically in Fig. 64, Curve A.
Fig. 64.
303The intensity of the radiation I after any time t is given by
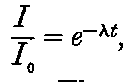
where λ is the radio-active constant.
The rate of decay of excited activity, like that of the activity of other radio-active products, is not appreciably affected by change of conditions. The rate of decay is independent of the concentration of the excited activity, and of the material of the body on which it is produced. It is independent also of the nature and pressure of the gas in which it decays. The rate of decay is unchanged whether the excited activity is produced on the body with or without an electric field.
The amount of excited activity produced on a body increases at first with the time, but reaches a maximum after an exposure of several days. An example of the results is given in the following table. In this experiment a rod was made the cathode in a closed vessel containing thoria. It was removed at intervals for the short time necessary to test its activity and then replaced.
| Time in hours | Current |
|---|---|
| 1·58 | 6·3 |
| 3·25 | 10·5 |
| 5·83 | 29 |
| 9·83 | 40 |
| 14·00 | 59 |
| 23·41 | 77 |
| 29·83 | 83 |
| 47·00 | 90 |
| 72·50 | 95 |
| 96·00 | 100 |
These results are shown graphically in Curve B, Fig. 64. It is seen that the decay and recovery curves may be represented approximately by the following equations.
For the decay curve A,
For the recovery curve B,
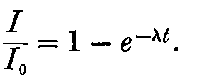
The two curves are thus complementary to one another; they are connected in the same way as the decay and recovery curves of Ur X, and are susceptible of a similar explanation.
304The amount of excited radio-activity reaches a maximum value when the rate of supply of fresh radio-active particles balances the rate of change of those already deposited.
180. Excited radio-activity produced by a short exposure. The initial portion of the recovery curve B, Fig. 64, is not accurately represented by the above equation. The activity for the first few hours increases more slowly than would be expected from the equation. This result, however, is completely explained in the light of later results. The writer[273] found that, for a short exposure of a body to the thorium emanation, the excited activity upon it after removal, instead of at once decaying at the normal rate, increased for several hours. In some cases the activity of the body increased to three or four times its original value in the course of a few hours and then decayed with the time at the normal rate.
For an exposure of 41 minutes to the emanation the excited activity after removal rose to three times its initial value in about 3 hours and then fell again at about the normal rate to half value in 11 hours.
With a longer time of exposure to the emanation, the ratio of the increase after removal is much less marked. For a day’s exposure, the activity after removal begins at once to diminish. In this case, the increase of activity of the matter deposited in the last few hours does not compensate for the decrease of activity of the active matter as a whole, and consequently the activity at once commences to decay. This increase of activity with time explains the initial irregularity in the recovery curve, for the active matter deposited during the first few hours takes some time to reach its maximum activity, and the initial activity is, in consequence, smaller than would be expected from the equation.
The increase of activity on a rod exposed for a short interval in the presence of the thorium emanation has been further investigated by Miss Brooks. The curve C in Fig. 65 shows the variation with time of the activity of a brass rod exposed for 10 minutes in the emanation vessel filled with dust-free air. The excited activity after removal increased in the course of 3·7 hours to five times its 305initial value, and afterwards decayed at the normal rate. The dotted line curve D represents the variation of activity to be expected if the activity decayed exponentially with the time. The explanation of this remarkable action is considered in detail in section 207.
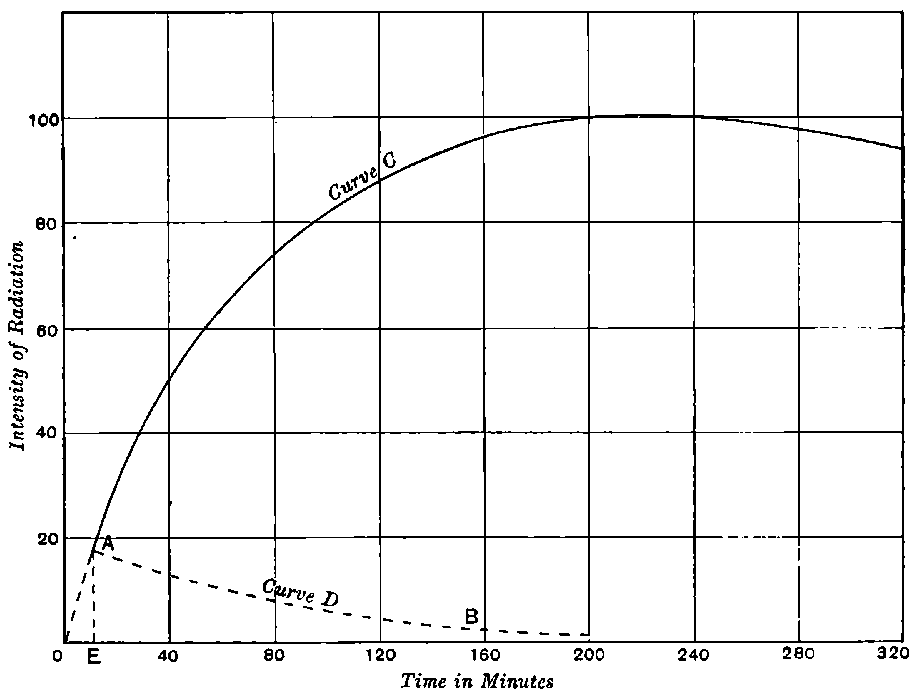
Fig. 65.
181. Effect of dust on the distribution of excited activity. Miss Brooks[274], working in the Cavendish Laboratory, observed that the excited activity due to the thorium emanation appeared in some cases on the anode in an electric field, and that the distribution of excited activity varied in an apparently capricious manner. This effect was finally traced to the presence of dust in the air of the emanation vessel. For example, with an exposure of 5 minutes the amount of excited activity to be observed on a rod depended on the time that the air had been allowed to remain undisturbed in the emanation vessel beforehand. The effect increased with the time of standing, and was a maximum after about 18 hours. The amount of excited activity obtained on the rod was then about 20 times as great as the amount observed for air freshly introduced. 306The activity of this rod did not increase after removal, but with fresh air, the excited activity, for an exposure of 5 minutes, increased to five or six times its initial value.
This anomalous behaviour was found to be due to the presence of dust particles in the air of the vessel, in which the bodies were made radio-active. These particles of dust, when shut up in the presence of the emanation, become radio-active. When a negatively charged rod is introduced into the vessel, a part of the radio-active dust is concentrated on the rod and its activity is added to the normal activity produced on the wire. After the air in the vessel has been left undisturbed for an interval sufficiently long to allow each of the particles of dust to reach a state of radio-active equilibrium, on the application of an electric field, all the positively charged dust particles will at once be carried to the negative electrode. The activity of the electrode at once commences to decay, since the decay of the activity of the dust particles on the wire quite masks the initial rise of the normal activity produced on the wire.
Part of the radio-active dust is also carried to the anode, and the proportion increases with the length of time during which the air has been undisturbed. The greatest amount obtained on the anode was about 60% of that on the cathode.
These anomalous effects were found to disappear if the air was made dust-free by passing through a plug of glass wool, or by application for some time of a strong electric field.
182. Decay of excited activity from radium. The excited activity produced on bodies by exposure to the radium emanation decays much more rapidly than the thorium excited activity. For short times of exposure[275] to the emanation the decay curve is very irregular. This is shown in Fig. 66.
It was found that the intensity of the radiation measured by the α rays decreased rapidly for the first 10 minutes after removal, but about 15 minutes after removal reached a value which remained nearly constant for an interval of about 20 minutes. It then decayed to zero, finally following an exponential law, the intensity falling to half value in about 28 minutes. With longer 307times of exposure, the irregularities in the curve are not so marked.
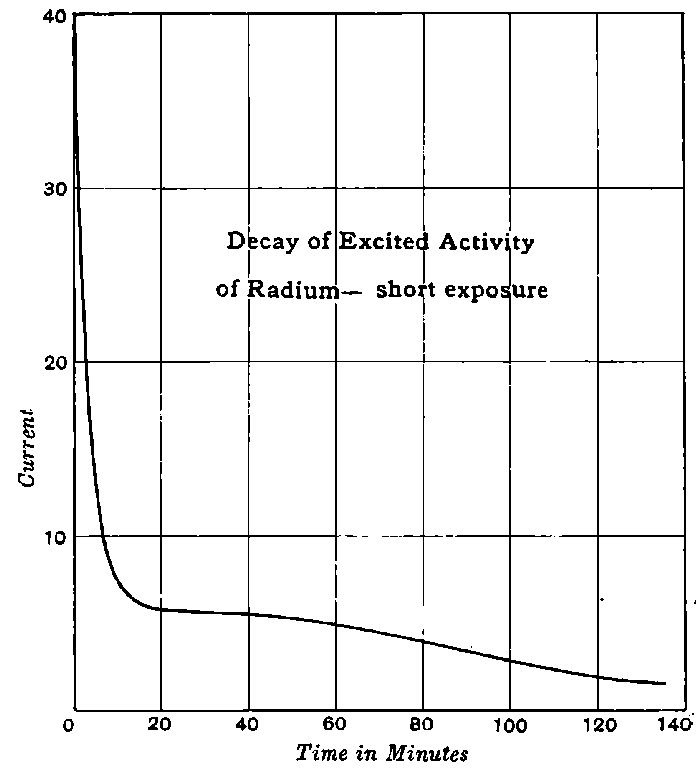
Fig. 66.
Miss Brooks has recently determined the decay curves of the excited activity of radium for different times of exposure, measured by the α rays. The results are shown in Fig. 67, where the initial ordinates represent the activity communicated to the body from different times of exposure to a constant supply of emanation. It will be observed that in all cases there is a sudden initial drop of activity, which becomes less marked with increasing time of exposure. The activity, several hours after removal, decreases exponentially in all cases, falling to half value in about 28 minutes.
Not only do the curves of variation of the excited activity after removal depend upon the time of exposure to the emanation, but they also depend upon whether the α or β and γ rays are used as a means of measurement. The curves obtained for the γ rays are identical with those from the β rays, showing that these two types 308of rays always occur together and in the same proportion. The curves measured by the β rays are very different, especially for the case of a short exposure to the emanation. This is clearly shown in Fig. 68, which gives the β and γ ray curves for exposures of 10 minutes, 40 minutes, and 1 hour, and also the limiting case of an exposure of 24 hours.
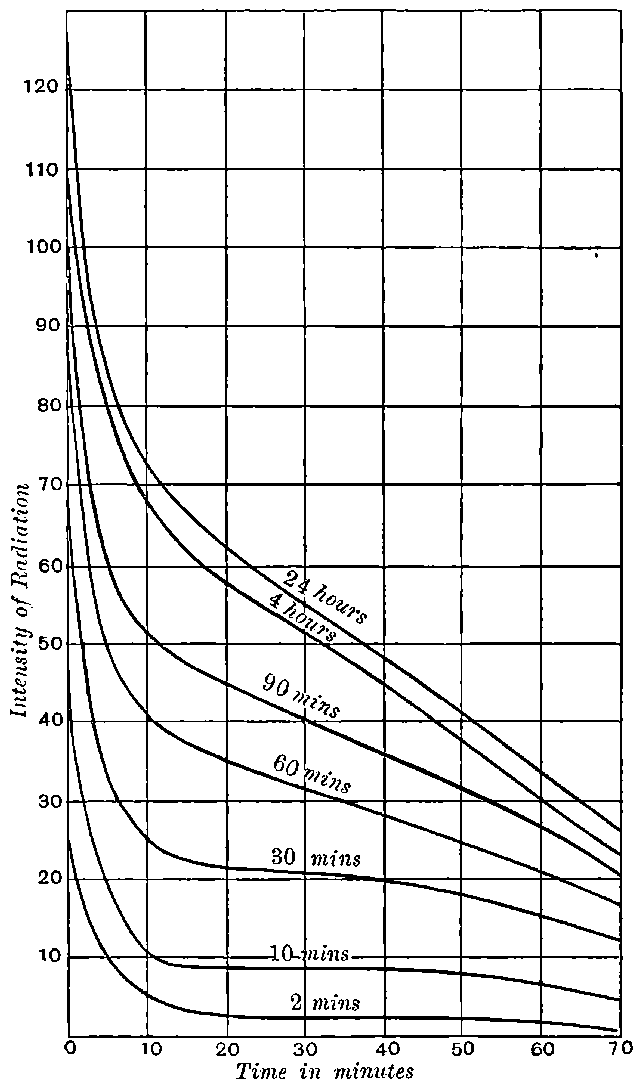
Fig. 67.
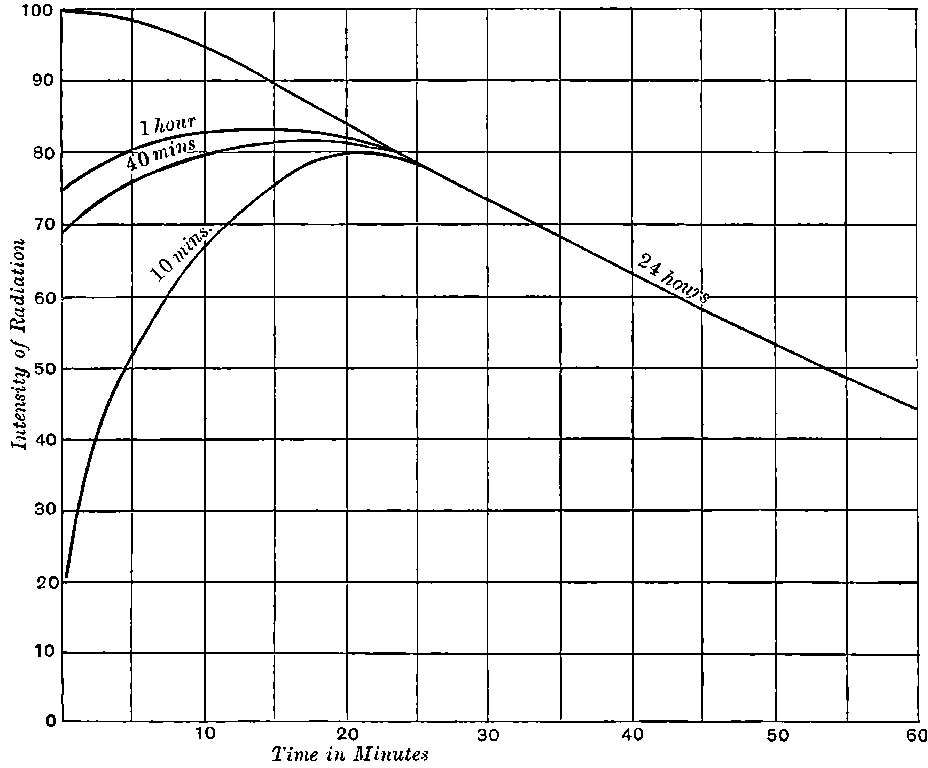
Fig. 68.
About 25 minutes after removal, the activity decays approximately at the same rate in each case. For convenience of representation, 309the ordinates of the curves were adjusted so that they all passed through a common point. We shall see later (chapter XI) that the rates of decay are not identically the same until several hours after removal; but, in the above figure, it is difficult to represent the slight variations. It will be observed that for the short exposure of 10 minutes the activity measured by the β rays is small at first but rises to a maximum in about 22 minutes, and then dies away with the time. The curve of decay of activity, measured by the β rays for a long exposure, does not show the rapid initial drop which occurs in all the α ray curves. Curie and Danne[276] made an investigation of the curves of decay of excited activity for different times of exposure to the radium emanation, 310but apparently did not take into account the fact that measurements made by the α and β rays give quite different curves of decay. Some of the family of curves, given in their paper, refer to the α rays and others to the β rays. They showed, however, the important fact that the curve of decay obtained by them for a long exposure (which is identical with the β ray curve) could be empirically expressed by an equation of the form
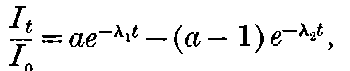
where I₀ is the initial intensity and It the intensity after any time t; λ1 = ¹⁄₂₄₂₀, λ2 = ¹⁄₁₈₆₀. The numerical constant a = 4·20. After an interval of 2·5 hours, the logarithmic decay curve is nearly a straight line, that is, the activity falls off according to an exponential law with the time, decreasing to half value in about 28 minutes.
The full explanation of this equation, and of the peculiarities of the various decay curves of the excited activity of radium, will be discussed in detail in chapter XI.
As in the case of the excited activity from thorium, the rate of decay of the excited activity from radium is for the most part independent of the nature of the body made active. Curie and Danne (loc. cit.) observed that the active bodies gave off an emanation itself capable of exciting activity in neighbouring bodies. This property rapidly disappeared, and was inappreciable 2 hours after removal. In certain substances like celluloid and caoutchouc, the decay of activity is very much slower than for the metals. This effect becomes more marked with increase of time of exposure to the emanation. A similar effect is exhibited by lead, but to a less marked degree. During the time the activity lasts, these substances continue to give off an emanation.
It is probable that these divergencies from the general law are not due to an actual change in the rate of decay of the true excited activity but to an occlusion of the emanation by these substances during the interval of exposure. After exposure the emanation gradually diffuses out, and thus the activity due to this occluded emanation and the excited activity produced by it decays very slowly with the time.
311 183. Active deposit of very slow decay. M. and Mme Curie[277] have observed that bodies which have been exposed for a long interval in the presence of the radium emanation do not lose all their activity. The excited activity at first decays rapidly at the normal rate, falling to half value in about 28 minutes, but a residual activity, which they state is of the order of ½0,000 of the initial activity, always remains. A similar effect was observed by Giesel. The writer has examined the variation of this residual activity, and has found that it increases for several years. The results are discussed in detail in chapter XI. It will there be shown that this active deposit of slow transformation contains the radio-active constituents present in polonium, radio-tellurium and radio-lead.
Fig. 69.
184. The excited activity from actinium. The emanation of actinium, like that of thorium and radium, produces excited activity on bodies, which is concentrated on the negative electrode in an electric field. Debierne[278] found that the excited activity 312decays approximately according to an exponential law, falling to half value in 41 minutes. Giesel[279] examined the rate of decay of the excited activity of “emanium”—which, we have seen, probably contains the same radio-active constituents as actinium—and found that it decayed to half value in 34 minutes. Miss Brooks[280] found that the curves of decay of the excited activity from Giesel’s emanium varied with the time of exposure to the emanation. The results are shown graphically in Fig. 69, for time exposures of 1, 2, 5, 10 and 30 minutes, and also for a long exposure of 21 hours. After 10 minutes the curves have approximately the same rate of decay. For convenience, the ordinates of the curves are adjusted to pass through a common point. For a very short exposure, the activity is small at first, but reaches a maximum about 9 minutes later and finally decays exponentially to zero.
The curve of variation of activity for a very short exposure has been determined accurately by Bronson; it is shown later in Fig. 83. He found that the decay of activity is finally exponential, falling to half value in 36 minutes.
The explanation of these curves is discussed in detail in chapter X, section 212.
185. Physical and chemical properties of the active deposit. On account of the slow decay of the activity of the active deposit from the thorium emanation, its physical and chemical properties have been more closely examined than the corresponding deposit from radium. It has already been mentioned that the active deposit of thorium is soluble in some acids. The writer[281] found that the active matter was dissolved off the wire by strong or dilute solutions of sulphuric, hydrochloric and hydrofluoric acids, but was only slightly soluble in water or nitric acid. The active matter was left behind when the solvent was evaporated. The rate of decay of activity was unaltered by dissolving the active matter in sulphuric acid, and allowing it to decay in the solution. In the experiment, the active matter was dissolved off an active platinum wire; then equal portions of the solutions were taken at definite intervals, evaporated down in 313a platinum dish, and the activity of the residue tested by the electric method. The rate of decay was found to be exactly the same as if the active matter had been left on the wire. In another experiment, an active platinum wire was made the cathode in a copper sulphate solution, and a thin film of copper deposited on it. The rate of decay of the activity was unchanged by the process.
A detailed examination of the physical and chemical properties of the active deposit of thorium has been made by F. von Lerch[282] and some important and interesting results have been obtained. A solution of the active deposit was prepared by dissolving the metal which had been exposed for some time in the presence of the thorium emanation. In most cases the active matter was precipitated with the metal. For example, an active copper wire was dissolved in nitric acid and then precipitated by caustic potash. The precipitate was strongly active. An active magnesium wire, dissolved in hydrochloric acid and then precipitated as phosphate, also gave an active precipitate. The activity of the precipitates decayed at the normal rate, i.e. the activity fell to half value in about 11 hours.
Experiments were also made on the solubility of the active deposit in different substances. A platinum plate was made active and then placed in different solutions, and the decrease of the activity observed. In addition to the acids already mentioned, a large number of substances were found to dissolve the active deposit to some extent. The active matter was however not dissolved to an appreciable extent in ether or alcohol. Many substances became active if added to the active solution and then precipitated. For example, an active solution of hydrochloric acid was obtained by dissolving the deposit on an active platinum wire. Barium chloride was then added and precipitated as sulphate. The precipitate was strongly active, thus suggesting that the active matter was carried down by the barium.
186. Electrolysis of solutions. Dorn showed that, if solutions of radiferous barium chloride were electrolysed, both electrodes became temporarily active, but the anode to a greater degree than the cathode. F. von Lerch has made a detailed examination of the action of electrolysis on a solution of the active deposit of 314thorium. The matter was dissolved off an active platinum plate by hydrochloric acid, and then electrolysed between platinum electrodes. The cathode was very active, but there was no trace of activity on the anode. The cathode lost its activity at a rate much faster than the normal. With an amalgamated zinc cathode on the other hand, the rate of decay was normal. When an active solution of hydrochloric acid was electrolysed with an electromotive force smaller than that required to decompose water, the platinum became active. The activity decayed to half value in 4·75 hours while the normal fall is to half value in 11 hours. These results point to the conclusion that the active matter is complex and consists of two parts which have different rates of decay of activity, and can be separated by electrolysis.
Under special conditions it was found possible to make the anode active. This was the case if the anion attached itself to the anode. For example, if an active hydrochloric solution was electrolysed with a silver anode, the chloride of silver formed was strongly active and its activity decayed at a normal rate. The amount of activity obtained by placing different metals in active solutions for equal times varied greatly with the metal. For example, it was found that if a zinc plate and an amalgamated zinc plate, which show equal potential differences with regard to hydrochloric acid, were dipped for equal times in two solutions of equal activity, the zinc plate was seven times as active as the other. The activity was almost removed from the solution in a few minutes by dipping a zinc plate into it. Some metals became active when dipped into an active solution while others did not. Platinum, palladium, and silver remained inactive, while copper, tin, lead, nickel, iron, zinc, cadmium, magnesium, and aluminium became active. These results strongly confirm the view that excited activity is due to a deposit of active matter which has distinctive chemical behaviour.
G. B. Pegram[283] has made a detailed study of the active deposits obtained by electrolysis of pure and commercial thorium salts. The commercial thorium nitrate obtained from P. de Haen gave, when electrolysed, a deposit of lead peroxide on the anode. This deposit was radio-active, and its activity decayed at the normal rate of the excited activity due to thorium. From solutions of 315pure thorium nitrate, no visible deposit was obtained on the anode, but it was, however, found to be radio-active. The activity decayed rapidly, falling to half value in about one hour. Some experiments were also made on the effect of adding metallic salts to thorium solutions and then electrolysing them. Anode and cathode deposits of the oxides or metals obtained in this way were found to be radio-active, but the activity fell to half value in a few minutes. The gases produced by electrolysis were radio-active, but this was due to the presence of the thorium emanation. The explanation of the results obtained by Pegram and von Lerch will be considered later in section 207. It will be shown that the active deposit of thorium contains two distinct substances which have different rates of transformation.
187. Effect of temperature. The activity of a platinum wire which has been exposed in the presence of the thorium emanation is almost completely lost by heating the wire to a white heat. Miss F. Gates[284] found that the activity was not destroyed by the intense heat, but manifested itself on neighbouring bodies. When the active wire was heated electrically in a closed cylinder, the activity was transferred from the wire to the interior surface of the cylinder in unaltered amount. The rate of decay of the activity was not altered by the process. By blowing a current of air through the cylinder during the heating, a part of the active matter was removed from the cylinder. Similar results were found for the excited activity due to radium.
F. von Lerch (loc. cit.) determined the amount of activity removed at different temperatures. The results are shown in the following table for a platinum wire excited by the thorium emanation[285].
| Temperature | Percentage of activity removed | |
| Heated 2 minutes | 800° C. | 0 |
| then „ ½ minute more | 1020° C. | 16 |
| „ „ ½ „ „ | 1260° C. | 52 |
| „ „ ½ „ „ | 1460° C. | 99 |
316The effect of heat on the volatilization of the active deposit of radium has been examined in detail by Curie and Danne. The interesting and important results obtained by them will be discussed in chapter XI, section 226.
188. Effect of variation of E.M.F. on amount of excited activity from thorium. It has been shown that the excited activity is confined to the cathode in a strong electric field. In weaker fields the activity is divided between the cathode and the walls of the vessel. This was tested in an apparatus[286] shown in Fig. 70.

Fig. 70.
A is a cylindrical vessel of 5·5 cms. diameter, B the negative electrode passing through insulating ends C, D. For a potential difference of 50 volts, most of the excited activity was deposited on the electrode B. For about 3 volts, half of the total excited activity was produced on the rod B, and half on the walls of the vessel. Whatever the voltage applied, the sum of the activities on the central rod and the walls of the cylinder was found to be a constant when a steady state was reached.
When no voltage was applied, diffusion alone was operative, and in that case about 13 per cent. of the total activity was on the rod B. The application of an electric field has thus no influence on the sum total of excited activity, but merely controls the proportion concentrated on the negative electrode.
A more detailed examination of the variation with strength of field of the amount on the negative electrode was made in a similar manner by F. Henning[287]. He found that in a strong electric field the amount of excited activity was practically independent of the diameter of the rod B, although the diameter varied between 317·59 mm. and 6·0 mms. With a small voltage, the amount on the negative electrode varied with its diameter. The curves showing the relation between the amount of excited activity and voltage are very similar in character to those obtained for the variation of the current through an ionized gas with the voltage applied.
The amount of excited activity reaches a maximum when all the active matter is removed from the gas as rapidly as it is formed. With weaker fields, a portion diffuses to the sides of the vessel, and produces excited activity on the positive electrode.
189. Effect of pressure on distribution of excited activity. In a strong electric field, the amount of excited activity produced on the cathode is independent of the pressure down to a pressure of about 10 mms. of mercury. In some experiments made by the writer[288], the emanating thorium compound was placed inside a closed cylinder about 4 cms. in diameter, through which passed an insulated central rod. The central rod was connected to the negative pole of a battery of 50 volts. When the pressure was reduced below 10 mms. of mercury, the amount of excited activity produced on the negative electrode diminished, and was a very small fraction of its original value at a pressure of ⅒ mm. Some excited activity was in this case found to be distributed over the interior surface of the cylinder. It may thus be concluded that at low pressures the excited activity appears on both anode and cathode, even in a strong electric field. The probable explanation of this effect is given in the next section.
Curie and Debierne[289] observed that when a vessel containing an emanating radium compound was kept pumped down to a low pressure, the amount of excited activity produced on the vessel was much reduced. In this case the emanation given off by the radium was removed by the pump with the other gases continuously evolved from the radium compound. On account of the very slow decay of activity of the emanation, the amount of excited activity produced on the walls of the vessel, in the passage of the emanation through it, was only a minute fraction of the amount produced when none of the emanation given off was allowed to escape.
318190. Transmission of excited activity. The characteristic property of excited radio-activity is that it can be confined to the cathode in a strong electric field. Since the activity is due to a deposit of radio-active matter on the electrified surface, the matter must be transported by positively charged carriers. The experiments of Fehrle[290] showed that the carriers of excited activity travel along the lines of force in an electric field. For example, when a small negatively charged metal plate was placed in the centre of a metal vessel containing an emanating thorium compound, more excited activity was produced on the sides and corners of the plate than at the central part.
A difficulty however arises in connection with the positive charge of the carrier. According to the view developed in section 136 and later in chapters X and XI, the active matter which is deposited on bodies and gives rise to excited activity, is itself derived from the emanation. The emanations of thorium and radium emit only α rays, i.e. positively charged particles. After the expulsion of an α particle, the residue, which is supposed to constitute the primary matter of the active deposit, should retain a negative charge, and be carried to the anode in an electric field. The exact opposite however is observed to be the case. The experimental evidence does not support the view that the positively charged α particles, expelled from the emanation, are directly responsible for the phenomena of excited activity; for no excited activity is produced in a body exposed to the α rays of the emanation, provided the emanation itself does not come in contact with it.
There has been a tendency to attach undue importance to this apparent discrepancy between theory and experiment. The difficulty is not so much to offer a probable explanation of the results as to select from a number of possible causes. While there can be little doubt that the main factor in the disintegration of the atom consists in the expulsion of an α particle carrying a positive charge, a complicated series of processes probably occurs before the residue of the atom is carried to the negative electrode. The experimental evidence suggests that one or more negative electrons of slow velocity escape from the atom at the same time as the 319particle. This is borne out by the recent discovery that the particle expelled from radium, freed from the ordinary β rays, and also from polonium, is accompanied by a number of slowly moving and consequently easily absorbed electrons. If two negative electrons escaped at the same time as the α particle, the residue would be left with a positive charge and would be carried to the negative electrode. There is also another experimental point which is of importance in this connection. In the absence of an electric field, the carriers remain in the gas for a considerable time and undergo their transformation in situ. There is also some evidence (section 227) that, even in an electric field, the carriers of the active deposit are not swept to the electrode immediately after the break up of the emanation, but remain some time in the gas before they gain a positive charge. It must be remembered that the atoms of the active deposit do not exist as a gas and by the process of diffusion would tend to collect together to form aggregates. These aggregates would act as small metallic particles, and, if they were electro-positive in regard to the gas, would gain a positive charge from the gas.
There can be little doubt that the processes occurring between the break up of the emanation and the deposit of the residue in the cathode in an electric field are complicated, and further careful experiment is required to elucidate the sequence of the phenomena.
Whatever view is taken of the process by which these carriers obtain a positive charge, there can be little doubt that the expulsion of an α particle with great velocity from the atom of the emanation must set the residue in motion. On account of the comparatively large mass of this residue, the velocity acquired will be small compared with that of the expelled α particle, and the moving mass will rapidly be brought to rest at atmospheric pressure by collision with the gas molecules in its path. At low pressures, however, the collisions will be so few that it will not be brought to rest until it strikes the boundaries of the vessel. A strong electric field would have very little effect in controlling the motion of such a heavy mass, unless it has been initially brought to rest by collision with the gas molecules. This would explain why the active matter is not deposited on the cathode at low pressures in an electric field. Some direct evidence of a 320process of this character, obtained by Debierne on examination of the excited activity produced by actinium, is discussed in section 192.
191. The following method has been employed by the writer[291] to determine the velocity of the positive carriers of excited activity of radium and thorium in an electric field. Suppose A and B (Fig. 71) are two parallel plates exposed to the influence of the emanation, which is uniformly distributed between them. If an alternating E.M.F. E₀ is applied between the plates, the same amount of excited activity is produced on each electrode. If, in series with the source of the alternating E.M.F., a battery of E.M.F. E1 less than E₀ is placed, the positive carrier moves in a stronger electric field in one half alternation than in the other. A carrier consequently moves over unequal distances during the two half alternations, since the velocity of the carrier is proportional to the strength of the electric field in which it moves. The excited activity will in consequence be unequally distributed over the two electrodes. If the frequency of alternation is sufficiently great, only the positive carriers within a certain small distance of one plate can be conveyed to it, and the rest, in the course of several succeeding alternations, are carried to the other plate.
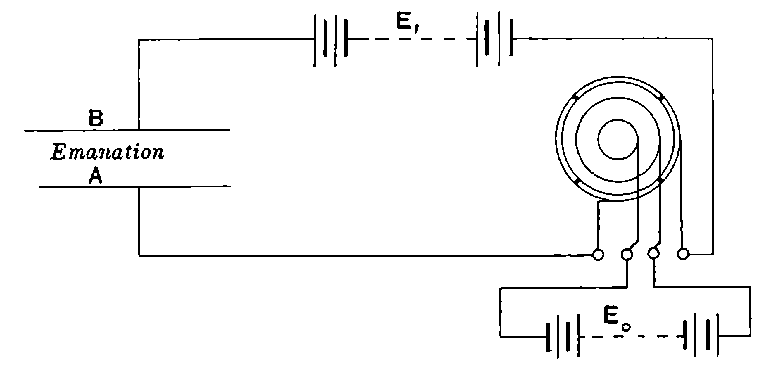
Fig. 71.
When the plate B is negatively charged, the E.M.F. between the plates is E₀ – E1, when B is positive the E.M.F. is E₀ + E1.
321Let
On the assumption that the electric field between the plates is uniform, and that the velocity of the carrier is proportional to the electric field, the velocity of the positive carrier towards B is
and, in the course of the next half alternation,
towards the plate A.
If x1 is less than d, the greatest distances x1, x2 passed over by the positive carrier during two succeeding half alternations is thus given by
Suppose that the positive carriers are produced at a uniform rate of q per second for unit distance between the plates. The number of positive carriers which reach B during a half alternation consists of two parts:
(1) One half of those carriers which are produced within the distance x1 of the plate B. This number is equal to
(2) All the carriers which are left within the distance x1 from B at the end of the previous half alternation. The number of these can readily be shown to be
The remainder of the carriers, produced between A and B during a complete alternation, will reach the other plate A in the course of succeeding alternations, provided no appreciable recombination 322takes place. This must obviously be the case, since the positive carriers travel further in a half alternation towards A than they return towards B during the next half alternation. The carriers thus move backwards and forwards in the changing electric field, but on the whole move towards the plate A.
The total number of positive carriers produced between the plates during a complete alternation is 2dqT. The ratio ρ of the number which reach B to the total number produced is thus given by
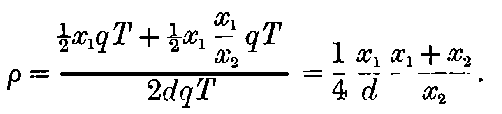
Substituting the values of x1 and x2, we find that
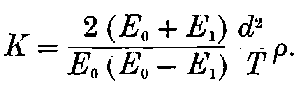
In the experiments, the values of E₀, E1, d, and T were varied, and the results obtained were in general agreement with the above equation.
The following were the results for thorium:
Plates 1·30 cms. apart.
| E₀ + E1 | E₀ – E1 | Alternations per second | ρ | K |
|---|---|---|---|---|
| 152 | 101 | 57 | ·27 | 1·25 |
| 225 | 150 | 57 | ·38 | 1·17 |
| 300 | 200 | 57 | ·44 | 1·24 |
Plates 2 cms. apart.
| E₀ + E1 | E₀ – E1 | Alternations per second | ρ | K |
|---|---|---|---|---|
| 273 | 207 | 44 | ·37 | 1·47 |
| 300 | 200 | 53 | ·286 | 1·45 |
The average mobility K deduced from a large number of experiments was 1·3 cms. per sec. per volt per cm. for atmospheric 323pressure and temperature. This velocity is about the same as the velocity of the positive ion produced by Röntgen rays in air, viz. 1·37 cms. per sec. The results obtained with the radium emanation were more uncertain than those for thorium on account of the distribution of some excited activity on the positive electrode. The values of the velocities of the carriers were however found to be roughly the same for radium as for thorium.
These results show that the carriers of the active deposit travel in the gas with about the same velocity as the positive or negative ions produced by the radiations in the gas. This indicates either that the active matter becomes attached to positive ions, or that the active matter itself, acquiring in some way a positive charge, collects a cluster of neutral molecules which travel with it.
192. Carriers of the excited activity from actinium and “emanium.” Giesel[292] observed that “emanium” gave off a large quantity of emanation, and that this emanation gave rise to a type of radiation which he termed the E rays. A narrow metal cylinder containing the active substance was placed with the open end downwards, about 5 cms. above the surface of a zinc sulphide screen. The screen was charged negatively to a high potential by an electric machine, and the cylinder connected with earth. A luminous spot of light was observed on the screen, which was brighter at the edge than at the centre. A conductor, connected with earth, brought near the luminous spot apparently repelled it. An insulator did not show such a marked effect. On removal of the active substance, the luminosity of the screen persisted for some time. This was probably due to the excited activity produced on the screen.
The results obtained by Giesel support the view that the carriers of excited activity of “emanium” have a positive charge. In a strong electric field the carriers travel along the lines of force to the cathode, and there cause excited activity on the screen. The movement of the luminous zone on the approach of a conductor is due to the disturbance of the electric field. 324Debierne[293] found that actinium also gave off a large amount of emanation, the activity of which decayed very rapidly with the time, falling to half value in 3·9 seconds.
This emanation produces excited activity on surrounding objects, and at diminished pressure the emanation produces a uniform distribution of excited activity in the enclosure containing the emanation. The excited activity falls to half value in 41 minutes.
Debierne observed that the distribution of excited activity was altered by a strong magnetic field. The experimental arrangement is shown in Fig. 71A. The active matter was placed at M, and two plates A and B were placed symmetrically with regard to the source. On the application of a strong magnetic field normal to the plane of the paper, the excited activity was unequally distributed between the plates A and B. The results showed that the carriers of excited activity were deviated by a magnetic field in the opposite sense to the cathode rays, i.e. the carriers were positively charged. In some cases, however, the opposite effect was obtained. Debierne considers that the excited activity of actinium is due to “ions activants,” the motion of which is altered by a magnetic field. Other experiments showed that the magnetic field acted on the “ions activants” and not on the emanation.
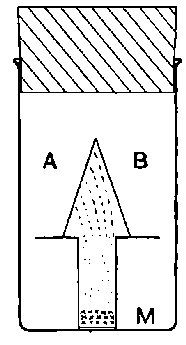
Fig. 71A.
The results of Debierne thus lead to the conclusion that the carriers of excited activity are derived from the emanation and are projected with considerable velocity. This result supports the view, advanced in section 190, that the expulsion of α particles from the emanation must set the part of the system left behind in rapid motion. A close examination of the mode of transference of the excited activity by actinium and the emanation substance is likely to throw further light on the processes which give rise to the deposit of active matter on the electrodes.
193. Introduction. We have seen in previous chapters that the radio-activity of the radio-elements is always accompanied by the production of a series of new substances with some distinctive physical and chemical properties. For example, thorium produces from itself an intensely radio-active substance, Th X, which can be separated from the thorium in consequence of its solubility in ammonia. In addition, thorium gives rise to a gaseous product, the thorium emanation, and also to another substance which is deposited on the surface of bodies in the neighbourhood of the thorium, where its presence is indicated by the phenomenon known as “excited activity.”
A close examination of the origin of these products shows that they are not produced simultaneously, but arise in consequence of a succession of changes originating in the radio-element. Thorium first of all gives rise to the product Th X. The Th X produces from itself the thorium emanation, and this in turn is transformed into a non-volatile substance. A similar series of changes is observed in radium, with the exception that there is no product in radium corresponding to the Th X in the case of thorium. Radium first of all produces an emanation, which, like thorium, is transformed into a non-volatile substance. In uranium only one product, Ur X, has been observed, for uranium does not give off an emanation and in consequence does not produce excited activity on bodies.
As a typical example of the evidence, from which it is deduced that one substance is the parent of another, we will consider the connection of the two products Th X and the thorium emanation. It has been shown (section 154) that after the separation of Th X 326from a thorium solution, by precipitation with ammonia, the precipitated thorium hydroxide has lost to a large extent its power of emanating. This cannot be ascribed to a prevention of escape of the emanation produced in it, for very little emanation is observed when a current of air is drawn through the hydroxide in a state of solution, when most of the emanation present would be carried off. On the other hand, the solution containing the Th X gives off a large quantity of emanation, showing that the power of giving off an emanation belongs to the product Th X. Now it is found that the quantity of emanation given off by the separated Th X decreases according to an exponential law with the time, falling to half value in four days. The rate of production of emanation thus falls off according to the same law and at the same rate as the activity of the Th X measured in the ordinary manner by the α rays. Now this is exactly the result to be expected if the Th X is the parent of the emanation, for the activity of Th X at any time is proportional to its rate of change, i.e., to the rate of production of the secondary type of matter by the emanation in consequence of a change in it. Since the rate of change of the emanation (half transformed in 1 minute) is very rapid compared with the rate of change of Th X, the amount of emanation present will be practically proportional to the activity of the Th X at any instant, i.e., to the amount of unchanged Th X present. The observed fact that the hydroxide regains its power of emanating in the course of time is due to the production of fresh Th X by the thorium, which in turn produces the emanation.
In a similar way, excited activity is produced on bodies over which the emanation is passed, and in amount proportional to the activity of the emanation, i.e., to the amount of the emanation present. This shows that the active deposit, which gives rise to the phenomenon of excited activity, is itself a product of the emanation. The evidence thus seems to be conclusive that Th X is the parent of the emanation and that the emanation is the parent of the deposited matter.
194. Chemical and Physical properties of the active products. Each of these radio-active products is marked by some distinctive chemical and physical properties which differentiate 327it from the preceding and succeeding products. For example, Th X behaves as a solid. It is soluble in ammonia, while thorium is not. The thorium emanation behaves as a chemically inert gas and condenses at a temperature of -120° C. The active deposit from the emanation behaves as a solid and is readily soluble in sulphuric and hydrochloric acids and is only slightly soluble in ammonia.
The striking dissimilarity which exists in many cases between the chemical and the physical properties of the parent matter and the product to which it gives rise is very well illustrated by the case of radium and the radium emanation. Radium is an element so closely allied in chemical properties to barium that, apart from a slight difference in the solubility of the chlorides and bromides, it is difficult to distinguish chemically between them. It has a definite spectrum of bright lines similar in many respects to the spectra of the alkaline earths. Like barium, it is non-volatile at ordinary temperature. On the other hand, the emanation which is continually produced from radium is a radio-active and chemically inert gas, which is condensed at a temperature of -150° C. Both in its spectrum and in the absence of definite chemical properties, it resembles the argon-helium group of inert gases, but differs from these gases in certain marked features.
The emanation must be considered to be an unstable gas which breaks down into a non-volatile type of matter, the disintegration being accompanied by the expulsion of heavy atoms of matter (α particles) projected with great velocity. This rate of breaking up is not affected by temperature over the considerable range which has been examined. After a month’s interval, the volume of the emanation has shrunk to a small portion of its initial value. But the most striking property of the emanation, which, as we shall see later (chapter XII), is a direct consequence of its radio-activity, is the enormous amount of energy emitted from it. The emanation in breaking up through its successive stages emits about 3 million times as much energy as is given out by the explosion of an equal volume of hydrogen and oxygen, mixed in the proper proportions to form water; and yet, in this latter chemical reaction more heat is emitted than in any other known chemical change.
328We have seen that the two emanations and the products Ur X, Th X lose their activity with the time according to a simple exponential law, and at a rate that is independent—as far as observation has gone—of the chemical and physical agents at our disposal. The time taken for each of these products to fall to half its value is thus a definite physical constant which serves to distinguish it from all other products.
On the other hand, the variation of the excited activity produced by these emanations does not even approximately obey such a law. The rate of decay depends not only on the time of exposure to the respective emanations, but also, in the case of radium, on the type of radiation which is used as a means of comparative measurement. It will be shown, in succeeding chapters, that the complexity of the decay is due to the fact that the matter in the active deposits undergoes several successive transformations, and that the peculiarities of the curves of decay, obtained under different conditions, can be explained completely on the assumption that two changes occur in the active deposit from both thorium and actinium and six in the active deposit from radium.
195. Nomenclature. The nomenclature to be applied to the numerous radio-active products is a question of great importance and also one of considerable difficulty. Since there are at least seven distinct substances produced from radium, and probably five from thorium and actinium, it is neither advisable nor convenient to give each a special name such as is applied to the parent elements. At the same time, it is becoming more and more necessary that each product should be labelled in such a way as to indicate its place in the succession of changes. This difficulty is especially felt in discussing the numerous changes in the active deposits from the different emanations. Many of the names attached to the products were given at the time of their discovery, before their position in the scheme of changes was understood. In this way the names Ur X, Th X were applied to the active residues obtained by chemical treatment of uranium and thorium. Since, in all probability, these substances are the first products of the two elements, it may be advisable to retain 329these names, which certainly have the advantage of brevity. The name “emanation” was originally given to the radio-active gas from thorium, and has since been applied to the similar gaseous products of radium and actinium.
Finding the name “radium emanation” somewhat long and clumsy, Sir William Ramsay[294] has recently suggested “ex-radio” as an equivalent. This name is certainly brief and is also suggestive of its origin; but at least six other ex-radios, whose parentage is as certain as that of the emanation, remain unnamed. A difficulty arises in applying the corresponding names ex-thorio, ex-actinio to the other gaseous products, for, unlike radium, the emanations of thorium and actinium are probably the second, not the first, disintegration product of the radio-elements in question. Another name thus has to be applied to the first product in these cases. It may be advisable to give a special name to the emanation, since it has been the product most investigated and was the first to be isolated chemically; but, on the other hand, the name “radium emanation” is historically interesting, and suggests a type of volatile or gaseous matter. Since the term “excited” or “induced” activity refers only to the radiations from the active body, a name is required for the radiating matter itself. The writer in the first edition of this book suggested the name “emanation X.”[295] This title was given from analogy to the names Ur X and Th X, to indicate that the active matter was product of the emanation. The name, however, is not very suitable, and, in addition, can only be applied to the initial product deposited, and not to the further products of its decomposition. It is very convenient in discussing mathematically the theory of successive changes to suppose that the deposited matter called A is changed into B, B into C, C into D, and so on. I have therefore discarded the name emanation X, and have used the terms radium A, radium B, and so on, to signify the successive products of the decomposition of the emanation of radium. A similar nomenclature is applied to thorium and actinium. This system of notation is elastic and simple, and I have found it of great convenience in the discussion of successive products. In 330speaking generally of the active matter, which causes excited activity, without regard to its constituents, I have used the term “active deposit.” The scheme of nomenclature employed in this book is clearly shown below:—
| Radium | Thorium | Uranium | Actinium |
| Radium emanation | Th X | Ur X | Actinium X |
| Radium A (Active) | Thorium emanation | Final product | Actinium emanation |
| Radium B (Active) | Thorium A (Active) | Actinium A (Active) | |
| Radium C (Active) | Thorium B (Active) | Actinium B (Active) | |
| Radium D (Active) | Thorium C (final) | Actinium C (final) | |
| &c. |
Each product on this scheme is the parent of the product below it. Since only two products have been observed in the active deposit of thorium and actinium, thorium C and actinium C respectively refer to their final inactive products. It will be shown in the next chapter that, as in the case of thorium, an intermediate product exists between actinium and its emanation. From analogy to the products Th X and Ur X, this substance is termed “actinium X.”
196. Theory of Successive Changes. Before considering the evidence from which these changes are deduced, the general theory of successive changes of radio-active matter will be considered. It is supposed that the matter A changes into B, B into C, C into D, and so on.
Each of these changes is supposed to take place according to the same law as a monomolecular change in chemistry, i.e., the number N of particles unchanged after a time t is given by
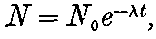
where N₀ is the initial number and λ the constant of the change.
Since dN/dt = -λN, the rate of change at any time is always proportional to the amount of matter unchanged. It has previously been pointed out that this law of decay of the activity of the radio-active products is an expression of the fact that the change is of the same type as a monomolecular chemical change.
331Suppose that P, Q, R represent the number of particles of the matter A, B, and C respectively at any time t. Let λ1, λ2, λ3 be the constants of change of the matter A, B, and C respectively.
Each atom of the matter A is supposed to give rise to one atom of the matter B, one atom of B to one of C, and so on.
The expelled “rays” or particles are non-radio-active, and so do not enter into the theory.
It is not difficult to deduce mathematically the number of atoms of P, Q, R, ... of the matter A, B, C, ... existing at any time t after this matter is set aside, if the initial values of P, Q, R, ... are given. In practice, however, it is generally only necessary to employ three special cases of the theory which correspond, for example, to the changes in the active deposit, produced on a wire exposed to a constant amount of radium emanation and then removed, (1) when the time of exposure is extremely short compared with the period of the changes, (2) when the time of exposure is so long that the amount of each of the products has reached a steady limiting value, and (3) for any time of exposure.
There is also another case of importance which is practically a converse of Case 3, viz. when the matter A is supplied at a constant rate from a primary source and the amounts of A, B, C are required at any subsequent time. The solution of this can, however, be deduced immediately from Case 3 without analysis.
197. Case 1. Suppose that the matter initially considered is all of one kind A. It is required to find the number of particles P, Q, R of the matter A, B, C respectively present after any time t.
Then
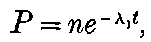
if n is the number of particles of A initially present. Now dQ, the increase of the number of particles of the matter B per unit time, is the number supplied by the change in the matter A, less the number due to the change of B into C, thus
Substituting in (1) the value of P in terms of n,
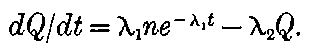
332The solution of this equation is of the form
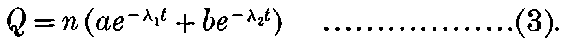
By substitution it is found that a = λ1/(λ2 – λ1).
Since Q = 0 when t = 0, b = -λ1(λ2 – λ1).
Thus
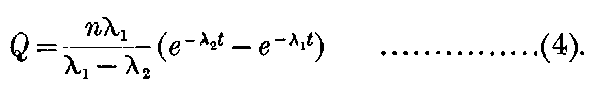
Substituting this value of Q in (2), it can readily be shown that
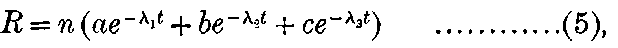
where
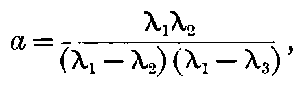
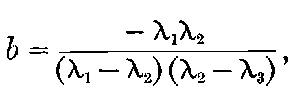
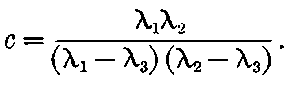
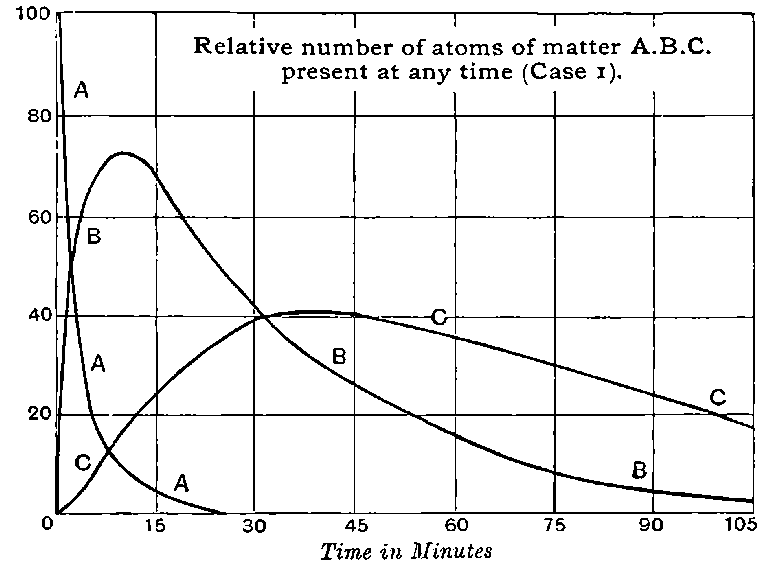
Fig. 72.
The variation of the values of P, Q, R with the time t, after removal of the source, is shown graphically in Fig. 72, curves A, B, and C respectively. In order to draw the curves for the practical case which will be considered later corresponding to the first three 333changes in radium A, the values of λ1, λ2, λ3 were taken as 3·85 × 10-3, 5·38 × 10-4, 4·13 × 10-4 respectively, i.e., the times required for each successive type of matter to be half transformed are about 3, 21, and 28 minutes respectively.
The ordinates of the curves represent the relative number of atoms of the matter A, B, and C existing at any time, and the value of n, the original number of atoms of the matter A deposited, is taken as 100. The amount of matter B is initially zero, and in this particular case, passes through a maximum about 10 minutes later, and then diminishes with the time. In a similar way, the amount of C passes through a maximum about 37 minutes after removal. After an interval of several hours the amount of both B and C diminishes very approximately according to an exponential law with the time, falling to half value after intervals of 21 and 28 minutes respectively.
198. Case 2. A primary source supplies the matter A at a constant rate and the process has continued so long that the amount of the products A, B, C, ... has reached a steady limiting value. The primary source is then suddenly removed. It is required to find the amounts of A, B, C, ... remaining at any subsequent time t.
In this case, the number n₀ of particles of A, deposited per second from the source, is equal to the number of particles of A which change into B per second, and of B into C, and so on. This requires the relation
where P₀, Q₀, R₀ are the maximum numbers of particles of the matter A, B, and C when a steady state is reached.
The values of P, Q, R at any time t after removal of the source are given by equations of the same form as (3) and (5) for a short exposure. Remembering the condition that initially
334it can readily be shown that
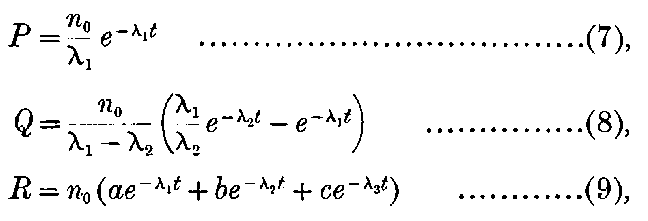
where
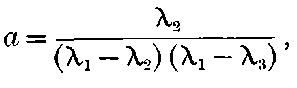
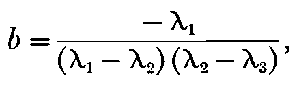
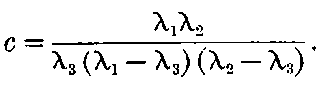
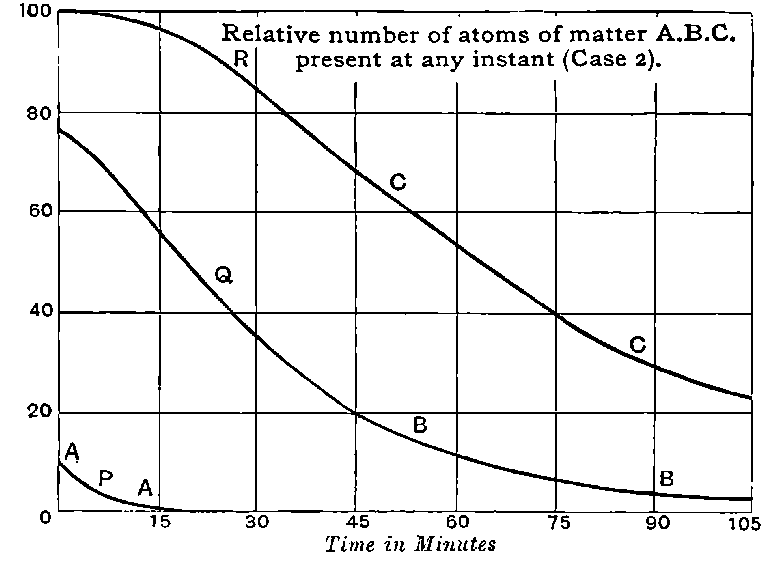
Fig. 73.
The relative numbers of atoms of P, Q, R existing at any time are shown graphically in Fig. 73, curves A, B, C respectively. The number of atoms R₀ is taken as 100 for comparison, and the values of λ1, λ2, λ3 are taken corresponding to the 3, 21, and 28-minute changes in the active deposit of radium. A comparison with Fig. 72 for a short exposure brings out very clearly the variation in the relative amounts of P, Q, R in the two cases. Initially the amount of R decreases very slowly. This is a result of the fact that the supply of C due to the breaking up of B at 335first, nearly compensates for the breaking up of C. The values of Q and R after several hours decrease exponentially, falling to half value in 28 minutes.
199. Case 3. Suppose that a primary source has supplied the matter A at a constant rate for any time T and is then suddenly removed. Required the amounts of A, B, C at any subsequent time.
Suppose that n₀ particles of the matter A are deposited each second. After a time of exposure T, the number of particles PT of the matter A present is given by
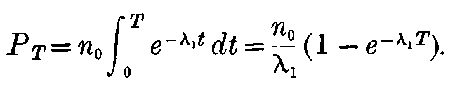
At any time t, after removal of the source, the number of particles P of the matter A is given by
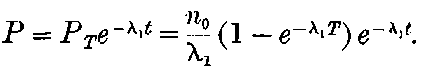
Consider the number of particles n₀dt of the matter A produced during the interval dt. At any later time t, the number of particles dQ of the matter B, which result from the change in A, is given (see equation 4) by
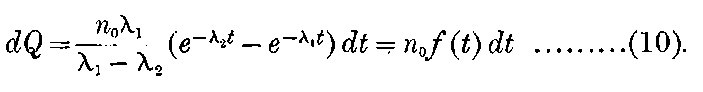
After a time of exposure T, the number of particles QT of the matter B present is readily seen to be given by
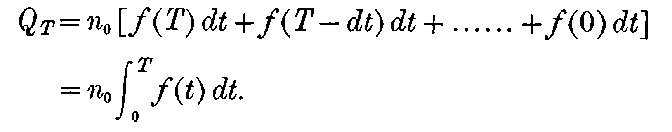
If the body is removed from the emanation after an exposure T, at any later time t the number of particles of B is in the same way given by
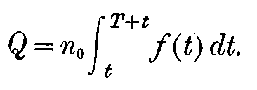
It will be noted that the method of deduction of QT and Q is independent of the particular form of the function f(t).
336Substituting the particular value of f(t) given in equation (10) and integrating, it can readily be deduced that
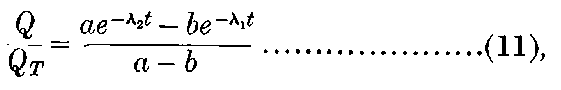
where
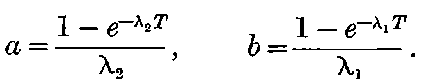
In a similar way, the number of particles R of the matter C present at any time can be deduced by substitution of the value of f(t) in equation (5). These equations are, however, too complicated in form for simple application to experiment, and will not be considered here.
200. Case 4. The matter A is supplied at a constant rate from a primary source. Required to find the number of particles of A, B, C at any subsequent time t, when initially A, B, C are absent.
The solution can be simply obtained in the following way. Suppose that the conditions of Case 2 are fulfilled. The products A, B, C are in radio-active equilibrium and let P₀, Q₀, R₀ be the number of particles of each present. Suppose the source is removed. The values of P, Q, R at any subsequent time are given by equations (7), (8) and (9) respectively. Now suppose the source, which has been removed, still continues to supply A at the same constant rate and let P1, Q1, R1 be the number of particles of A, B, C again present with the source at any subsequent time. Now we have seen, that the rate of change of any individual product, considered by itself, is independent of conditions and is the same whether the matter is mixed with the parent substance or removed from it. Since the values of P₀, Q₀, R₀ represent a steady state where the rate of supply of each kind of matter is equal to its rate of change, the sum of the number of particles A, B, C present at any time with the source, and in the matter from which it was removed, must at all times be equal to P₀, Q₀, R₀, ..., that is
337This must obviously be the case, for otherwise there would be a destruction or creation of matter by the mere process of separation of the source from its products; but, by hypothesis, neither the rate of supply from the source, nor the law of change of the products, has been in any way altered by removal.
Substituting the values of P, Q, R from equations (7), (8), and (9), we obtain
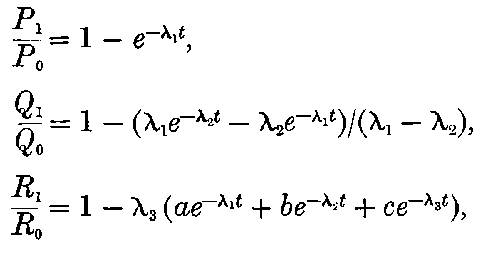
where a, b, and c have the values given after equation (9). The curves representing the increase of P, Q, R, are thus, in all cases, complementary to the curves shown in Fig. 73. The sum of the ordinates of the two curves of rise and decay at any time is equal to 100. We have already seen examples of this in the case of the decay and recovery curves of Ur X and Th X.
201. Activity of a mixture of products. In the previous calculations we have seen how the number of particles of each of the successive products varies with the time under different conditions. It is now necessary to consider how this number is connected with the activity of the mixture of products.
If N is the number of particles of a product, the number of particles breaking up per second is λN, where λ is the constant of change. If each particle of each product, in breaking up, emits one α particle, we see that the number of α particles expelled per second from the mixture of products at any time is equal to λ1P + λ2Q + λ3R + ..., where P, Q, R, ... are the numbers of particles of the successive products A, B, C, .... Substituting the values of P, Q, R already found from any one of the four cases previously considered, the variation of the number of α particles expelled per second with the time can be determined.
The ideal method of measuring the activity of any mixture of radio-active products would be to determine the number of α 338or β particles expelled from it per second. In practice, however, this is inconvenient and also very difficult experimentally.
Certain practical difficulties arise in endeavouring to compare the activity of one product with another. We shall see later that, in many cases, all of the successive products do not emit α rays. Some give out β and γ rays alone, while there are several “rayless” products, that is, products which do not emit either α, β, or γ rays. In the case of radium, for example, radium A gives out only α rays, radium B no rays at all, while radium C gives out α, β, and γ rays.
In practice, the relative activity of any individual product at any time is usually determined by relative measurements of the saturation ionization current produced between the electrodes of a suitable testing vessel.
Let us consider, for example, the case of a product which gives out only α rays. The passage of the α particles through the gas produces a large number of ions in its path. Since the α particles from any individual product are projected with the same average velocity under all conditions, the relative amount of the ionization produced per second in the testing vessel serves as an accurate means of determining the variation of its activity. No two products, however, emit α particles with the same average velocity. We have seen that the rays from some products are more readily stopped in the gas than others. Thus the relative saturation current, due to two different products in a testing vessel, does not serve as an accurate method of comparing the relative number of α particles expelled per second. The ratio of the currents will in general depend upon the distance between the plates of the testing vessel, and, unless the relative ionization due to the average α particle from the two products is known from other data, the comparison of the currents can, at best, be only an approximate guide to the relative number of α particles escaping into the gas.
202. Some examples will now be considered to show how the factors, above considered, influence the character of the curves of activity obtained under different experimental conditions. For the purpose of illustration, we shall consider the variation after removal of the excited activity on a body exposed for different times to a constant supply of the radium emanation. The active 339deposit on removal consists in general of a mixture of the products radium A, B, and C. The nature of the rays from each product, the time for each product to be transformed, and the value of λ are tabulated below for convenience:—
| Product | Rays | T. | λ (sec-1) |
|---|---|---|---|
| Radium A | α rays | 3 min. | 3·85 × 10-3 |
| Radium B | no rays | 21 min. | 5·38 × 10-4 |
| Radium C | α, β, γ rays | 28 min. | 4·13 × 10-4 |
Since only the product C gives rise to β and γ rays, the activity measured by either of these types of rays will be proportional to the amount of C present at any time, i.e. to the value of R at any time. For a long exposure, the variation of activity with time measured by the β and γ rays will thus be represented by the upper curve CC of Fig. 73, where the ordinates represent activity. This curve will be seen to be very similar in shape to the experimental curve for a long exposure which is given in Fig. 68.
Since radium B does not give out rays, the number of α particles expelled from the active deposit per second is proportional to λ1P + λ3R. The activity measured by the α rays, using the electrical method, is thus proportional at any time to λ1P + Kλ3R, where K is a constant which represents the ratio of the number of ions, produced in the testing vessel, by an α particle from C compared with that from an α particle emitted by A.
It will be seen later that, for this particular case, K is nearly unity. Taking K = 1, the activity at any time after removal is proportional to λ1P + λ3R.
Case 1. We shall first consider the activity curve for a short exposure to the radium emanation. The relative values of P, Q, and R at any time corresponding to this case are graphically shown in Fig. 74. The activity measured by the α rays at any time will be the sum of the activities due to A and C separately.
Let curve AA (Fig. 74) represent the activity due to A. This decreases exponentially, falling to half value in 3 minutes. In order to show the small activity due to C clearly in the Figure, the activity due to A is plotted after an interval of 6 minutes, when the activity has been reduced to 25 per cent. of its maximum 340value. The activity due to C is proportional to λ3R, and in order to represent the activity due to C to the same scale as A, it is necessary to reduce the scale of the ordinates of curve CC in Fig. 72 in the ratio λ3/λ1.
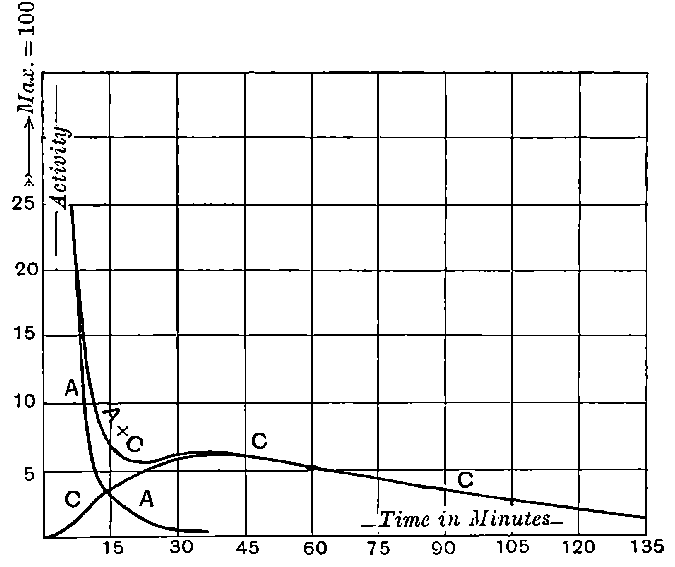
Fig. 74.
The activity due to C is thus represented by the curve CCC, Fig. 74. The total activity is thus represented by a curve A + C whose ordinates are the sum of the ordinates of A and C.
This theoretical activity curve is seen to be very similar in its general features to the experimental curve shown in Fig. 66, where the activity from a very short exposure is measured by the α rays.
Case 2. The activity curve for a long exposure to the emanation will now be considered. The activity after removal of A and C is proportional to λ1P + λ3R, where the values of P and R are graphically shown in Fig. 75 by the curves AA, CC. Initially after removal, λ1P₀ = λ3R₀, since A and C are in radio-active equilibrium, and the same number of particles of each product break up per second. The activity due to A alone is shown in curve AA, Fig. 75. The activity decreases exponentially, falling to half value in 3 minutes. The activity due to C at any time is proportional 341to R, and is initially equal to that of A. The activity curve due to C is thus represented by the curve CC, which is the same curve as the upper curve CC of Fig. 73. The activity of A and C together is represented by the upper curve A + C (Fig. 75), where the ordinates are equal to the sum of the ordinates of the curves A and C. This theoretical curve is seen to be very similar in shape to the experimental curve (Fig. 67) showing the decay of activity of the active deposit from a long exposure measured by the α rays.
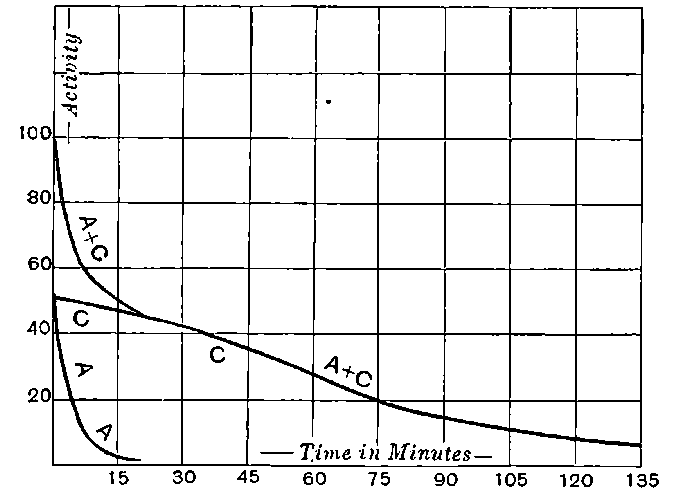
Fig. 75.
203. Effect of a rayless change on the activity curves. Certain important cases occur in the analysis of radio-active changes, when one of the products does not give rise to rays and so cannot be detected directly. The presence of this rayless change can, however, be readily observed by the variations which occur in the activity of the succeeding product.
Let us consider, for example, the case where the inactive matter A, initially all of one kind, changes into the matter B which gives out rays. The inactive matter A is supposed to be transformed according to the same law as the radio-active products. Let λ1, λ2 be the constants of the change of A and B respectively. If n is the number of particles of A, initially present, we see from 342the equation (4), section 197, that the number of particles of the matter B present at any time is given by
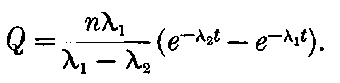
Differentiating and equating to zero, it is seen that the value of Q passes through a maximum at a time T given by the equation
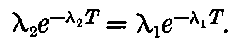
For the sake of illustration, we shall consider the variation of the activity of the active deposit of thorium, due to a very short exposure to the emanation. Thorium A gives out no rays, and thorium B gives out α, β, and γ rays, while thorium C is inactive.
The matter A is half transformed in 11 hours, and B is half transformed in 55 minutes. The value of λ1 = 1·75 x 10-5(sec.)-1 and λ2 = 2·08 x 10-4(sec.)-1.
The activity of the mixture of products A + B is due to B alone, and will, in consequence, be always proportional to the amount of B present, that is, to the value of Q.
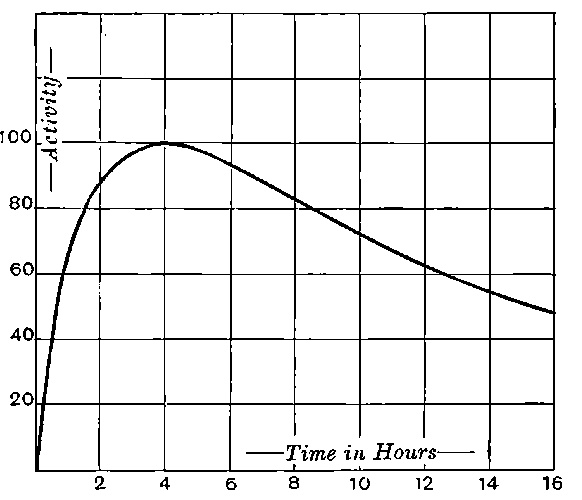
Fig. 76.
The variation of activity with time is shown graphically in Fig. 76. The activity rises from zero to a maximum in 220 minutes and then decays, finally decreasing, according to an exponential law, with the time, falling to half value in 11 hours.
343This theoretical curve is seen to agree closely in shape with the experimental curve (Fig. 65), which shows the variation of the activity of the active deposit of thorium, produced by a short exposure in presence of the emanation.
There are several points of interest in connection with an activity curve of this character. The activity, some hours after removal, decays according to an exponential law, not at the rate of the product B, from which the activity rises, but at the same rate as the first rayless transformation. This will also be the case if the rayless product has a slower rate of change than the succeeding active product. Given an activity curve of the character of Fig. 76, we can deduce from it that the first change is not accompanied by rays and also the period of the two changes in question. We are, however, unable to determine from the curve which of the periods of change refers to the rayless product. It is seen that the activity curve is unaltered if the values of λ1, λ2, that is, if the periods of the products are interchanged, for the equation is symmetrical in λ1, λ2. For example, in the case of the active deposit of thorium, without further data it is impossible to decide whether the period of the first change has a value of 55 minutes or 11 hours. In such cases the question can only be settled by using some physical or chemical means in order to separate the product A from B, and then testing the rate of decay of their activity separately. In practice, this can often be effected by electrolysis or by utilizing the difference in volatility of the two products. If now a product is separated from the mixture of A and B which loses its activity according to an exponential law, falling to half value in 55 minutes (and such is experimentally observed), we can at once conclude that the active product B has the period of 55 minutes.
The characteristic features of the activity curve shown in Fig. 76 becomes less marked with increase of the time of exposure of a body to the emanation, that is, when more and more of B is mixed with A at the time of removal. For a long time of exposure, when the products A and B are in radio-active equilibrium, the activity after removal is proportional to Q, where
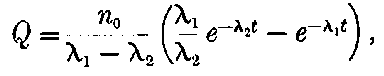
344(see equation 8, section 198). The value of Q, in this case, does not increase after removal, but at once commences to diminish. The activity, in consequence, decreases from the moment of removal, but more slowly than would be given by an exponential law. The activity finally decays exponentially, as in the previous case, falling to half value in 11 hours.
In the previous case we have discussed the activity curve obtained when both the active and inactive product have comparatively rapid rates of transformation. In certain cases which arise in the analysis of the changes in actinium and radium, the rayless product has a rate of change extremely slow compared with that of the active product. This corresponds to the case where the active matter B is supplied from A at a constant rate. The activity curve will thus be identical in form with the recovery curves of Th X and Ur X, that is, the activity I at any time t will be represented by the equation
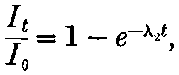
where I₀ is the maximum value of the activity and λ2 the constant of change of B.
204. In this chapter we have considered the variation with time, under different conditions, of the number of atoms of the successive products, when the period and number of the changes are given. It has been seen that the activity curves to be expected under various conditions can be readily deduced from the simple theory. In practice, however, the investigator has been faced with the much more difficult inverse problem of deducing the period, number, and character of the products, by analysis of the activity curves obtained under various conditions.
In the case of radium, where at least seven distinct changes occur, the problem has been one of considerable difficulty, and a solution has only been possible by devising special physical and chemical methods of isolation of some of the products.
We shall see later that two rayless changes occur in radium and actinium and one in thorium. It is at first sight a very striking fact that the presence of a substance which does not emit rays can be detected, and its properties investigated. This is only possible when the rayless product is transformed into another 345substance which emits rays; for the variation of the activity of the latter may be such as to determine not only the period but also the physical and chemical properties of the parent product. In the two following chapters the application of the theory of successive changes will be shown to account satisfactorily for the complicated processes occurring in the radio-elements.
205. In the last chapter the mathematical theory of successive changes has been considered. The results there obtained will now be applied to explain the radio-active phenomena observed with uranium, thorium, actinium, radium, and their products.
It has been shown in sections 127 and 129 that a radio-active constituent Ur X can be separated from uranium by several different processes. The activity of the separated Ur X decays with the time, falling to half value in about 22 days. At the same time the uranium, from which the Ur X has been separated, gradually regains its lost activity. The laws of decay of Ur X and of the recovery of the lost activity of the uranium are expressed by the equations
and
where λ is the radio-active constant of Ur X. The substance Ur X is produced from uranium at a constant rate, and the constant radio-activity observed in uranium represents a state of equilibrium, where the rate of production of new active matter is balanced by the rate of change of the Ur X already produced.
The radio-active processes occurring in uranium present several points of difference from the processes occurring in thorium and radium. In the first place, uranium does not give off an emanation, and in consequence does not produce any excited activity on bodies. So far only one active product Ur X has been observed in uranium. This active product Ur X differs from Th X and the emanations, 347inasmuch as the radiation from it consists almost entirely of β rays. This peculiarity of the radiations from Ur X initially led to some confusion in the interpretation of observations on Ur X and the uranium from which it had been separated. When examined by the photographic method, the uranium freed from Ur X showed no activity, while the Ur X possessed it to an intense degree. With the electric method, on the other hand, the results obtained were exactly the reverse. The uranium freed from Ur X showed very little loss of activity, while the activity of the Ur X was very small. The explanation of these results was given by Soddy[296] and by Rutherford and Grier[297]. The α rays of uranium are photographically almost inactive, but produce most of the ionization in the gas. The β rays, on the other hand, produce a strong photographic action, but very little ionization compared with the α rays. When the Ur X is separated from the uranium, the uranium does not at first give out any β rays. In the course of time fresh Ur X is produced from the uranium, and β rays begin to appear, gradually increasing in intensity until they reach the original value shown before the separation of the Ur X.
In order to determine the recovery curves of uranium after the separation of Ur X, it was thus necessary to measure the rate of increase of the β rays. This was done by covering the uranium with a layer of aluminium of sufficient thickness to absorb all the α rays, and then measuring the ionization due to the rays in an apparatus similar to Fig. 17.
Uranium has not yet been obtained inactive when tested by the electric method. Becquerel[298] has stated that he was able to obtain inactive uranium, but in his experiments the uranium was covered with a layer of black paper, which would entirely absorb the α rays. There is no evidence that the α radiation of uranium has been altered either in character or amount by any chemical treatment. The α rays appear to be inseparable from the uranium, and it will be shown later that thorium and radium as well as uranium also possess a non-separable activity consisting entirely of α rays. The changes occurring in uranium must then be 348considered to be of two kinds, (1) the change which gives rise to the α rays and the product Ur X, (2) the change which gives rise to the β rays from Ur X.
The possibility of separating the Ur X, which gives rise to the β rays of uranium, shows that the α and β rays are produced quite independently of one another, and by matter of different chemical properties.
Following the general considerations discussed in section 136 we may suppose that every second some of the atoms of uranium—a very minute fraction of the total number present will suffice—become unstable and break up, expelling an α particle with great velocity. The uranium atom, minus one α particle, becomes the atom of the new substance, Ur X. This in turn is unstable and breaks up with the expulsion of the β particle and the appearance of a γ ray.
The changes occurring in uranium are graphically shown in Fig. 77.
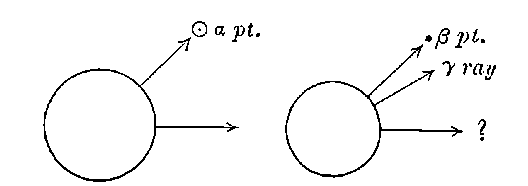
Fig. 77.
On this view the α ray activity of uranium should be an inherent property of the uranium, and should be non-separable from it by physical or chemical means. The β and γ ray activity of uranium is a property of Ur X, which differs in chemical properties from the parent substance and can at any time be completely removed from it. The final product, after the decay of Ur X, is so slightly active that its activity has not yet been observed. We shall see later (chapter XIII.) that there is some reason to believe that the changes in uranium do not end at this point but continue through one or more stages, finally giving rise to radium, or in other words that radium is a product of the disintegration of the uranium atom. Meyer and Schweidler[299], in a recent paper, state that the activity due to uranium preparations increases somewhat in a 349closed vessel. On removing the uranium no residual activity, however, was observed. They consider that this effect may be due to a very short-lived emanation emitted by uranium.
206. Effect of crystallization on the activity of uranium. Meyer and Schweidler[300] recently observed that uranium nitrate, after certain methods of treatment, showed remarkable variations of its activity, measured by the β rays. The α ray activity, on the other hand, was unaltered. Some uranium nitrate was dissolved in water and then shaken up with ether, and the ether fraction drawn off. The early experiments of Crookes showed that, by this method, the uranium in the ether portion was photographically inactive. This is simply explained by supposing that the uranium X is insoluble in ether, and consequently remained behind in the water fraction. The ether fraction gradually regained its β ray activity at the normal rate to be expected if Ur X was produced by the uranium at a constant rate, for it recovered half its final activity in about 22 days. Some of the uranium in the water fraction was crystallized and placed under an electroscope. The β ray activity fell rapidly at first to half its value in the course of four days. The activity then remained constant, and no further change was observed over an interval of one month. Other experiments were made with crystals of uranium nitrate, which had not been treated with ether. The nitrate was dissolved in water and a layer of crystals separated. The β ray activity of these crystals fell rapidly at first, the rate varying somewhat in different experiments, but reached a minimum value after about five days. The β ray activity then rose again at a slow rate for several months.
The rapid drop of activity of the crystals seemed, at first sight, to indicate that crystallization was able in some way to alter the activity of uranium.
Dr Godlewski, working in the laboratory of the writer, repeated the work of Meyer and Schweidler, and obtained results of a similar character, but the initial drop of activity was found to vary both in rate and amount in different experiments. These results were at first very puzzling and difficult to explain, for the mother liquor, left behind after removal of the crystals, did not show the 350corresponding initial rise, which would be expected if the variation of activity were due to the partial separation of some new product of uranium.
The cause of this effect was, however, rendered very evident by a few well-considered experiments made by Godlewski. The uranium nitrate was dissolved in hot water in a flat dish, and allowed to crystallize under the electroscope. Up to the moment of crystallization the β ray activity remained constant, but as soon as the crystals commenced to form at the bottom of the solution the β ray activity rapidly rose in the course of a few minutes to five times the initial value. After reaching a maximum, the activity very gradually decreased again to the normal value. If, however, the plate of crystals was reversed, the β ray activity was found at first to be much smaller than the normal, but increased as fast as that of the other side diminished.
The explanation of this effect is simple. Ur X is very soluble in water and, at first, does not crystallize with the uranium, but remains in the solution, and, consequently, when the crystallization commences at the bottom of the vessel the upper layer of liquid becomes richer in uranium X. Since the β rays arise only from the product Ur X and not from the uranium itself, and the Ur X is mostly confined to the upper layer, a much greater proportion of the β rays escape than if the Ur X were uniformly distributed throughout the thick layer of uranium. When the amount of water added is just sufficient to supply the water of crystallization, the Ur X in the upper layer of crystals gradually diffuses back through the mass and, in consequence, the activity of the upper surface diminishes and of the lower surface rises. A similar explanation applies to the effects observed by Meyer and Schweidler. The water fraction, left behind after treatment with ether, contained all the Ur X. The first layer of crystals formed in it contained some Ur X, and this was for the most part confined to the top layer of crystals. The amount of β rays at first diminished owing to the gradual diffusion of the Ur X from the surface. In the first experiment, the amount of Ur X present was in radio-active equilibrium with the uranium, and, after the initial drop, the β ray activity remained constant. In the second experiment, the gradual rise is due to the fact that the crystals of uranium first formed 351contained less than the equilibrium amount of Ur X. After falling to a minimum, the β ray activity, in consequence, slowly rose again to the equilibrium value.
These effects exhibited by uranium are of great interest, and illustrate in a striking manner the difference in properties of Ur X and the uranium. The gradual diffusion of the Ur X throughout the mass of crystals is noteworthy. By measurements of the variation with time of the β ray activity, it should be possible to deduce its rate of diffusion into the crystallized mass.
207. Analysis of the active deposit. The radio-active processes occurring in thorium are far more complicated than those in uranium. It has already been shown in chapter vi that a radio-active product Th X is continuously produced from the thorium. This Th X breaks up, giving rise to the radio-active emanation. The emanation produces from itself a type of active matter which is deposited on the surface of bodies, where it gives rise to the phenomena of excited or induced activity. This active deposit possesses some distinctive chemical and physical properties which distinguish it from the emanation and the Th X. We have seen (section 180) that the rate at which the active deposit loses its activity depends upon the time of exposure of the body made active to the emanation. The explanation of the activity curves for different time of exposure will now be considered.
The curve of variation of activity for a short exposure of 10 minutes has already been given in Fig. 65. The activity is small at first but increases rapidly with the time; it passes through a maximum about 4 hours later, and finally decays exponentially with the time, falling to half value in 11 hours. This remarkable effect can be explained completely[301] if it be supposed that the active deposit consists of two distinct substances. The matter initially deposited from the emanation, which will be called thorium A, is supposed to be changed into thorium B. Thorium A is transformed according to the ordinary exponential 352law, but the change is not accompanied by any ionizing rays. In other words, the change from A to B is a “rayless” change. On the other hand, B breaks up into C with the accompaniment of all three kinds of rays. On this view the activity of the active deposit at any time represents the amount of the substance B present, since C is inactive or active to a very minute extent.
If the variation of the activity imparted to a body exposed for a short interval in the presence of the thorium emanation, is due to the fact that there are two successive changes in the deposited matter A, the first of which is a “rayless” change, the activity It at any time t after removal should be proportional to the number Qt of particles of the matter B present at that time. Now, from equation (4) section 197, it has been shown that
The value of Qt passes through a maximum QT at the time T when
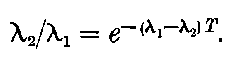
The maximum activity IT is proportional to QT and
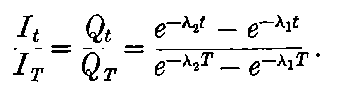
It will be shown later that the variation with time of the activity, imparted to a body by a short exposure, is expressed by an equation of the above form. It thus remains to fix the values of λ1, λ2. Since the above equation is symmetrical with regard to λ1, λ2, it is not possible to settle from the agreement of the theoretical and experimental curve which value of λ refers to the first change. The curve of variation of activity with time is unaltered if the values of λ1 and λ2 are interchanged.
It is found experimentally that the activity 5 or 6 hours after removal decays very approximately according to an exponential law with the time, falling to half value in 11 hours. This is the normal rate of decay of thorium for all times of exposure, provided measurements are not begun until several hours after the removal of the active body from the emanation.
353This fixes the value of the constants of one of the changes. Let us assume for the moment that this gives the value of λ1.
Since the maximum activity is reached after an interval T = 220 minutes (see Fig. 65), substituting the values of λ1 and T in the equation, the value of λ2 comes out to be
This value of λ2 corresponds to a change in which half the matter is transformed in 55 minutes.
Substituting now the values of λ1, λ2, T, the equation reduces to
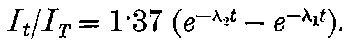
The agreement between the results of the theoretical equation and the observed values is shown in the following table:
| Time in minutes | Theoretical value of It/IT | Observed value of It/IT |
|---|---|---|
| 15 | ·22 | ·23 |
| 30 | ·38 | ·37 |
| 60 | ·64 | ·63 |
| 120 | ·90 | ·91 |
| 220 | 1·00 | 1·00 |
| 305 | ·97 | ·96 |
After 5 hours the activity decreased nearly exponentially with the time, falling to half value in 11 hours.
It is thus seen that the curve of rise of activity for a short exposure is explained very satisfactorily on the supposition that two changes occur in the deposited matter, of which the first is a rayless change.
Further data are required in order to fix which of the time constants of the changes refers to the first change. In order to settle this point, it is necessary to isolate one of the products of the changes and to examine the variation of its activity with time. If, for example, a product can be separated whose activity decays to half value in 55 minutes, it would show that the second change is the more rapid of the two. Now Pegram[302] has examined the radio-active products obtained by electrolysis of thorium solutions. 354The rates of decay of the active products depended upon conditions, but he found that, in several cases, rapidly decaying products were obtained whose activity fell to half value in about 1 hour. Allowing for the probability that the product examined was not completely isolated by the electrolysis, but contained also a trace of the other product, this result would indicate that the last change which gives rise to rays is the more rapid of the two.
This point is very clearly brought out by some recent experiments of Miss Slater[303], who has made a detailed examination of the effect of temperature on the active deposit of thorium.
A platinum wire was made active by exposure for a long interval to the thorium emanation, and then heated for a few minutes to any desired temperature by means of the electric current. The wire, while being heated, was surrounded by a lead cylinder in order that any matter driven off from it should be collected on its surface. The decay of activity both of the wire and of the lead cylinder was then tested separately. After heating to a dull red heat, no sensible diminution of the activity was observed at first, but the rate of decay of the activity on the wire was found to be more rapid than the normal. The activity of the lead cylinder was small at first but increased to a maximum after about 4 hours and then decayed at the normal rate with the time.
These results are to be expected if some thorium A is volatilized from the wire; for the rise of activity on the lead cylinder is very similar to that observed on a wire exposed for a short time in the presence of the thorium emanation, i.e., under the condition that only thorium A is initially present.
On heating the wire above 700° C. the activity was found to be reduced, showing that some thorium B had also been removed. By heating for a few minutes at about 1000° C. nearly all the thorium A was driven off. The activity on the wire then decayed exponentially with the time, falling to half value in about 1 hour. After heating for a minute at about 1200° C. all the activity was removed. These results show that thorium A is more volatile than B, and that the product which gives out rays, viz. thorium B, has a period of about 55 minutes.
Another series of experiments was made, in which an active 355aluminium disc was placed in an exhausted tube, and exposed to the cathode ray discharge. Under these conditions, a part of the activity of the disc was removed. When the disc was made the anode, the loss of activity was usually 20 to 60 per cent. for half-an-hour’s exposure. If the disc was made the cathode, the loss was much greater, amounting to about 90 per cent. in 10 minutes. Part of the active matter removed from the disc was collected on a second disc placed near it. This second disc on removal lost its activity at a far more rapid rate than the normal. The rate of decay on the first disc was also altered, the activity sometimes even increasing after removal. These results indicate that, in this case, the apparent volatility of the products is reversed. Thorium B is driven off from the disc more readily than thorium A. The rates of decay obtained under different conditions were satisfactorily explained by supposing that the surfaces of the discs after exposure to the discharge were coated with different proportions of thorium A and B.
The escape of thorium B from the disc under the influence of the discharge seems rather to be the result of an action similar to the well-known “sputtering” of electrodes than to a direct influence of temperature.
The results obtained by von Lerch[304] on the electrolysis of a solution of the active deposit also admit of a similar interpretation. Products were obtained on the electrodes of different rates of decay, losing half their activity in times varying from about 1 hour to 5 hours. This variation is due to the admixture of the two products in different proportions. The evidence, as a whole, thus strongly supports the conclusion that the active deposit from thorium undergoes two successive transformations as follows:
(1) A “rayless” change for which λ1 = 1·75 × 10-5, i.e., in which half the matter is transformed in 11 hours;
(2) A second change giving rise to α, β and γ rays, for which λ2 = 2·08 × 10-4, i.e., in which half the matter is transformed in 55 minutes[305].
356It is, at first sight, a somewhat unexpected result that the final rate of decay of the active deposit from thorium gives the rate of change not of the last product itself, but of the preceding product, which does not give rise to rays at all.
A similar peculiarity is observed in the decay of the excited activity of actinium, which is discussed in section 212.
For a long exposure in the presence of a constant supply of thorium emanation, the equation expressing the variation of activity with time is found from equation (8), section 198,
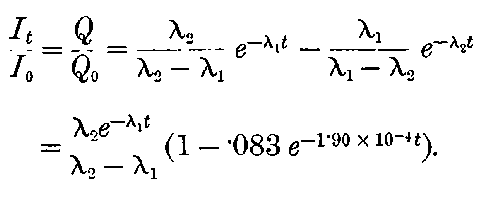
About 5 hours after removal the second term in the brackets becomes very small, and the activity after that time will decay nearly according to an exponential law with the time, falling to half value in 11 hours. For any time of exposure T, the activity at time t after the removal (see equation 11, section 199) is given by
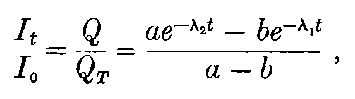
where I₀ is the initial value of the activity, immediately after removal, and
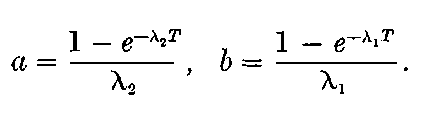
By variation of T the curves of variation of activity for any time of exposure can be accurately deduced from the equation, when the values of the two constants λ1, λ2 are substituted. Miss Brooks[306] has examined the decay curves of excited activity for thorium for different times of exposure and has observed a substantial agreement between experiment and theory.
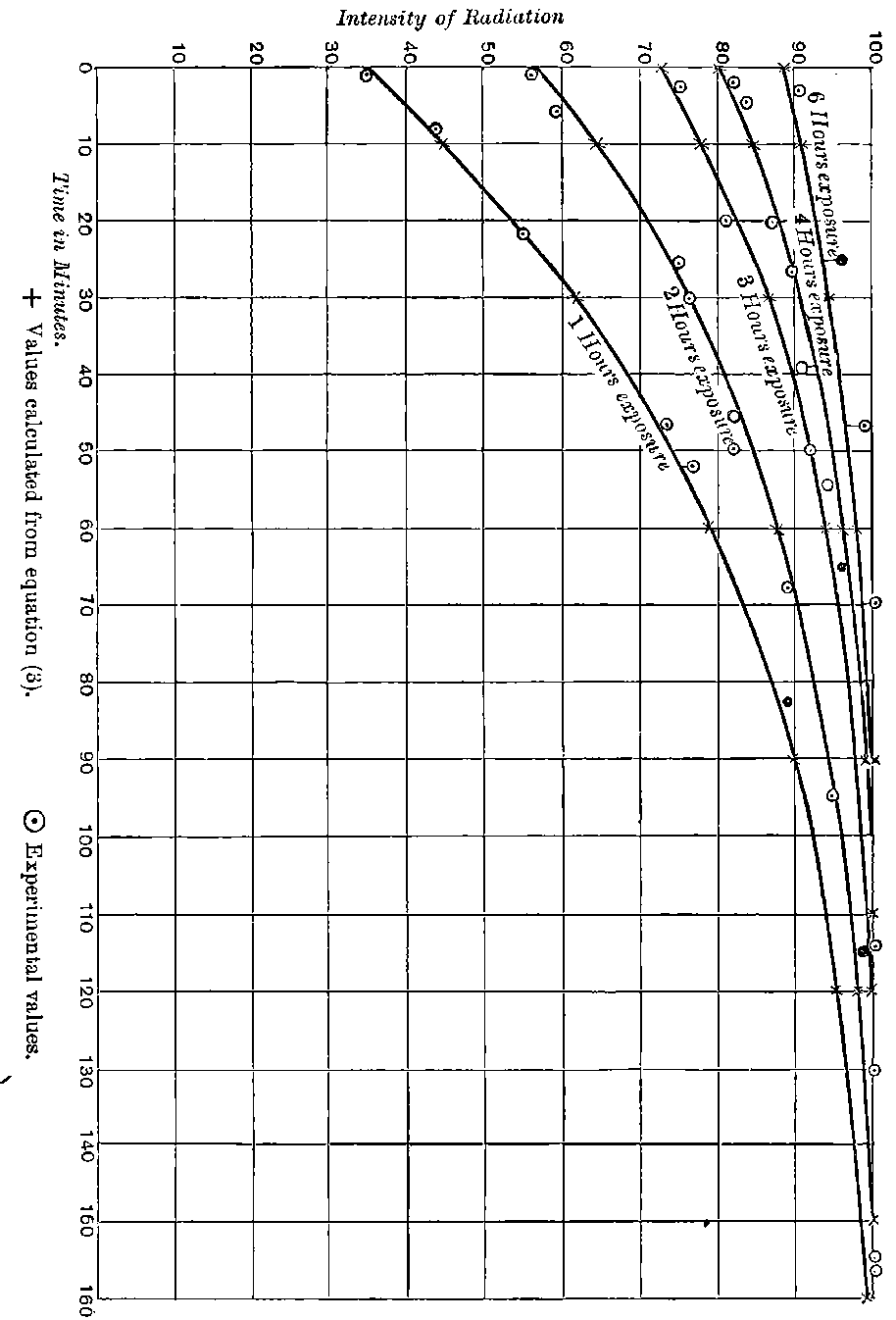
Fig. 78.
The results are shown graphically in Fig. 78. The maximum 357value of the activity is, for each time of exposure, taken as 100. The theoretical and observed values are shown in the Figure.
358 208. Analysis of the decay and recovery curves of Th X. The peculiarities of the initial portions of the decay and recovery curves of Th X and thorium respectively (Curves A and B, Fig. 47, p. 221), will now be considered. It was shown that when the Th X was removed from the thorium by precipitation with ammonia, the radiation increased about 15 per cent. during the first day, passed through a maximum, and then fell off according to an exponential law, decreasing to half value in four days. At the same time the activity of the separated hydroxide decreased for the first day, passed through a minimum, and then slowly increased again, rising to its original value after the lapse of about one month.
When a thorium compound is in a state of radio-active equilibrium, the series of changes in which Th X, the emanation, and thorium A and B are produced, go on simultaneously. Since a state of equilibrium has been reached for each of these products, the amount of each product changing in unit time is equal to the amount of that product supplied from the preceding change in unit time. Now the matter Th X is soluble in ammonia, while thorium A and B are not. The Th X is thus removed from the thorium by precipitation with ammonia, but A and B are left behind with the thorium. Since the active deposit is produced from the emanation, which in turn arises from Th X, on the removal of the parent matter Th X, the radiation due to this active deposit will decay, since the rate of production of fresh matter no longer balances its own rate of change. Disregarding the initial irregularity in the decay curve of the active deposit, its activity will have decayed to half value in about 11 hours, and to one quarter value at the end of 22 hours. As soon, however, as the Th X has been separated, new Th X is produced in the thorium compound. The activity of this new Th X is not, however, sufficient to compensate at first for the loss of activity due to the change in the active deposit, so that, as a whole, the activity will at first decrease, then pass through a minimum, then increase again.
The correctness of this point of view has been tested by Rutherford and Soddy[307] as follows: If the precipitated thorium hydroxide 359after the removal of Th X is put through a series of precipitations with ammonia at short intervals, the Th X is removed almost as fast as it is formed, and, at the same time, the activity of thorium B in the thorium decays.
The following table indicates the results obtained. A portion of the precipitated hydroxide was removed after each series of precipitations and its activity tested in the usual way.
| Activity of hydroxide per cent. | |
|---|---|
| After 1 precipitation | 46 |
| After 3 precipitations at intervals of 24 hours | 39 |
| After 3 more precipitations at intervals of 24 hours and 3 at intervals of 8 hours | 22 |
| After 3 more each of 8 hours | 24 |
| After 6 more each of 4 hours | 25 |
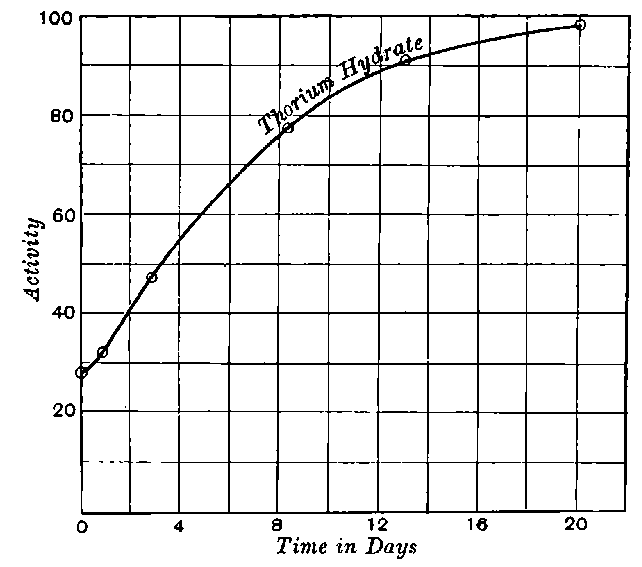
Fig. 79.
The differences in the last three numbers are not significant, for it is difficult to make accurate comparisons of the activity of thorium compounds which have been precipitated under slightly different conditions. It is thus seen that as a result of successive precipitations, the activity is reduced to a minimum of about 25 per cent. The recovery curve of the activity of this 23 times precipitated 360hydroxide is shown in Fig. 79. The initial drop in the curve is quite absent, and the curve, starting from the minimum, is practically identical with the curve shown in Fig. 48, which gives the recovery curve of thorium hydroxide after the first two days. This residual activity—about 25 per cent. of the maximum—is non-separable from the thorium by any chemical process that has been tried.
The initial rise of activity of Th X, after it has been separated, will now be considered. In all cases it was found that the activity of the separated Th X had increased about 15 per cent. at the end of 24 hours, and then steadily decayed, falling to half value in about four days.
This peculiarity of the Th X curve follows, of necessity, from the considerations already advanced to explain the drop in the recovery curve. As soon as the Th X is separated, it at once produces from itself the emanation, and this in turn produces thorium A and B. The activity due to B at first more than compensates for the decay of activity of the Th X itself. The total activity thus increases to a maximum, and then slowly decays to zero according to an exponential law with the time. The curve expressing the variation of the activity of the separated Th X with time can be deduced from the theory of successive changes already considered in chapter IX. In the present case there are four successive changes occurring at the same time, viz. the change of Th X into the emanation, of the emanation into thorium A, of A into B, and of B into an inactive product. Since, however, the change of the emanation into thorium A (about half changed in one minute) is far more rapid than the changes occurring in Th X or thorium A and B, for the purposes of calculation it may be assumed without serious error that the Th X changes at once into the active deposit. The 55 minute change will also be disregarded for the same reason.
Let λ1 and λ2 be the constants of decay of activity of Th X and of thorium A respectively. Since the activity of Th X and of thorium A falls to half value in 4 days and 11 hours respectively, the value of λ1 = ·0072 and of λ2 = ·063, where 1 hour is taken as the unit of time.
The problem reduces to the following: Given the matter A 361(thorium X) all of one kind, which changes into B (thorium B), find the activity of A and B together at any subsequent time. This corresponds to Case I. (section 197). The amount Q of B at any time T is given by
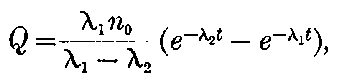
and the activity I at any time of the two together is proportional to λ1P + Kλ2Q, where K is the ratio of the ionization of B compared with that of A.
Then
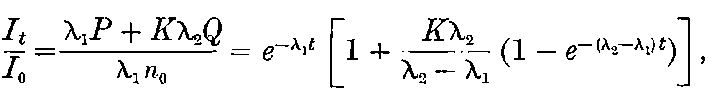
where I₀ is the initial activity due to n₀ particles of Th X.
By comparison of this equation with the curve of variation of the activity of Th X with time, shown in Fig. 47, it is found that K is almost ·44. It must be remembered that the activity of the emanation and Th X are included together, so that the activity of thorium B is about half of the activity of the two preceding products.
The calculated values of It/I₀ for different values of t are shown in the second column of the following table, and the observed values in the third column.
| Time | Theoretical value | Observed value |
|---|---|---|
| 0 | 1·00 | 1·00 |
| ·25 days | 1·09 | — |
| ·5 „ | 1·16 | — |
| 1 „ | 1·15 | 1·17 |
| 1·5 „ | 1·11 | — |
| 2 „ | 1·04 | — |
| 3 „ | ·875 | ·88 |
| 4 „ | ·75 | ·72 |
| 6 „ | ·53 | ·53 |
| 9 „ | ·315 | ·295 |
| 13 „ | ·157 | ·152 |
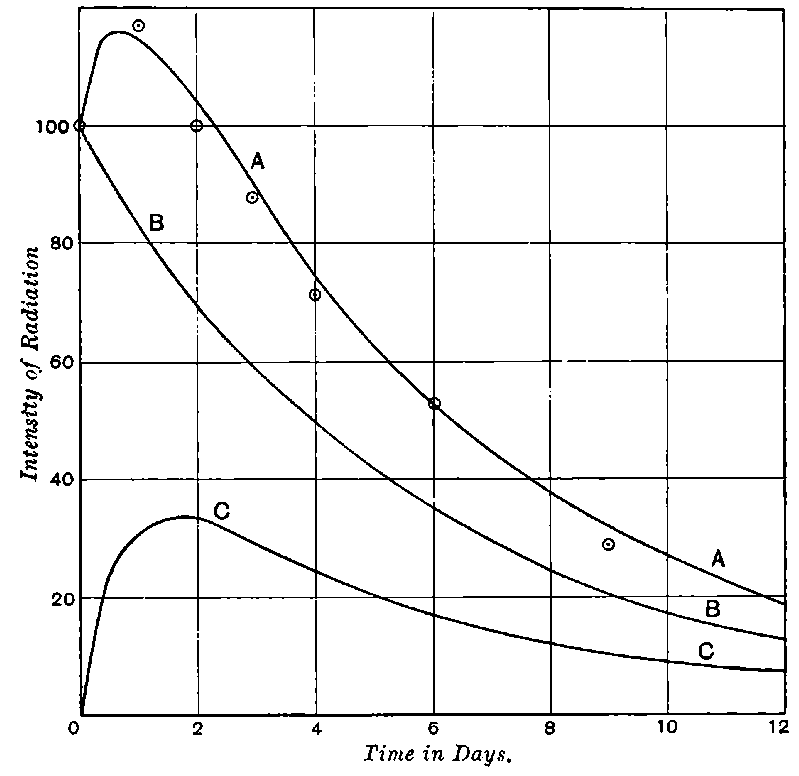
Fig. 80.
The theoretical and observed values thus agree within the limit of error in the measurements. The theoretical curve is shown in Curve A, Fig. 80 (with the observed points marked, for comparison). The curve B shows the theoretical curve of the decay of the activity of Th X and the emanation, supposing there is no further change into the active deposit. Curve C shows the difference curve between the curves A and B, i.e. the proportion of the activity at different times due to the active deposit. The activity due to the latter thus rises to a maximum about two days after removal of the Th X, and then decays with the time at the same rate as the Th X itself, i.e. the activity falls to half value every four days. When t exceeds four days, the term
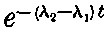
in the theoretical equation is very small.
363The equation of decay after this time is therefore expressed by
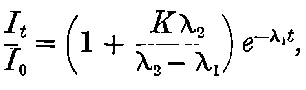
i.e. the activity decays according to an exponential law with the time.
209. Radiations from Thorium products. It has been shown in the last section that the activity of thorium, by successive precipitations with ammonia, is reduced to a limiting value of almost 25 per cent. of the initial activity. This “non-separable activity” consists of α rays, the β and γ rays being altogether absent. According to the disintegration theory, this is an expression of the fact that the initial break-up of the thorium atom is accompanied only by the expulsion of α particles. We have seen in section 156 that the thorium emanation also gives out only α rays. In the active deposit, thorium A gives out no rays, while thorium B emits all three types of rays.
Some hours after separation, Th X gives out α, β, and γ rays, but the appearance of β and γ rays is probably due to the thorium B associated with it. The β and γ ray activity of Th X is much reduced if a current of air is continuously aspirated through a solution of Th X to remove the emanation. It seems likely that if the emanation could be removed as fast as it was formed, so as to prevent the formation of thorium B in its mass, Th X itself would give out only α rays: but, on account of the rapid rate of change of the thorium emanation, it is difficult to realize this experimentally.
210. Transformation products of Thorium. The transformation products of thorium and the rays emitted by them are graphically shown below (Fig. 81).
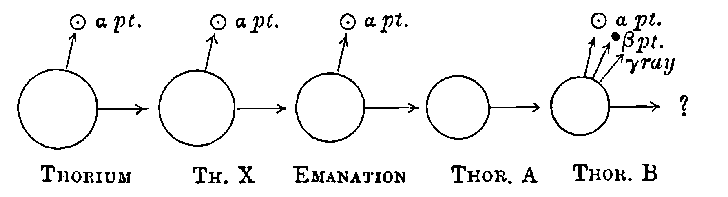
Fig. 81.
364A table of the transformation products of thorium is shown below, with some of their physical and chemical properties.
| Product | Time to be half transformed | λ (sec)-1 | Radiations | Physical and chemical properties |
|---|---|---|---|---|
| Thorium | α rays | Insoluble in ammonia | ||
| Th. X | 4 days | 2·00 × 10-6 | α rays | Soluble in ammonia |
| Emanation | 54 secs. | 1·28 × 10-2 | α rays | Inert gas, condenses -120° C. |
| Thorium A | 11 hours | 1·75 × 10-5 | no rays | Soluble in strong acids. Volatile at a white heat. B can be separated from A by electrolysis and by difference of volatility. |
| Thorium B | 55 mins. | 2·1 × 10-4 | α, β, γ rays | Same |
| ? | — | — | — | - |
211. Transformation products of Actinium. It has previously been pointed out (sections 17 and 18) that the actinium of Debierne and the emanium of Giesel contain the same radio-active constituent. Both give out a short-lived emanation which imparts activity to the surface of bodies. Recently, thanks to Dr Giesel of Braunschweig, preparations of “emanium” have been placed on the market, and most of the investigations that are described later have been made with this substance.
Actinium X. Actinium and thorium are very closely allied in radio-active properties. Both emit an emanation which is rapidly transformed, but the rate of change of the actinium emanation is still more rapid than that of thorium, the activity decreasing to half value in 3·7 seconds. Miss Brooks[308] has analysed the active deposit from the emanation of actinium, and has shown that two successive changes occur in it, very similar in character to those observed in the active deposit of thorium. It thus seemed 365probable, from analogy, that an intermediate product, corresponding to Th X in thorium, would be found in actinium[309]. Recent work has verified this supposition. Giesel[310] and Godlewski[311] independently observed that a very active substance could be separated from “emanium,” very similar in chemical and physical properties to Th X in thorium. This product will, from analogy, be called “actinium X.” The same method, which was used by Rutherford and Soddy to separate Th X from thorium, is also effective in separating actinium X from actinium. After precipitation of the active solution with ammonia, actinium X is left behind in the filtrate. After evaporation and ignition, a very active residue remains. At the same time, the precipitated actinium loses a large proportion of its activity.
Giesel observed the separation of an active product, using a fluorescent screen to detect the radiations. A very complete examination of the product actinium X has been made by Godlewski in the laboratory of the writer.
After separation of actinium X, the activity, whether measured by the α or β rays, increases about 15 per cent. during the first day, and afterwards decays exponentially with the time, falling to half value in 10·2 days. The activity of the separated actinium was small at first but steadily increased with the time, reaching a practical maximum after an interval of sixty days. After the first day, the decay and recovery curves of activity are complementary to one another. The curves of rise and decay are shown graphically in Fig. 82, curves I and II respectively.
Godlewski observed that a solution of actinium, freed from actinium X, gave out very little emanation, while a solution of actinium X gave off the emanation in large quantity. The amount of emanation from the solution was measured by observing the activity produced in a testing vessel, similar to that shown in Fig. 51, when a constant current of air was passed through the solution. The emanating power of actinium X decreased exponentially with the time at the same rate as that at which the actinium X lost its activity. At the same time the actinium solution increased 366in emanating power, reaching its original value after about 60 days. The behaviour of actinium and thorium is thus quite analogous, and the explanation advanced to explain the decay and recovery curves of thorium applies equally well to the corresponding curves of actinium.
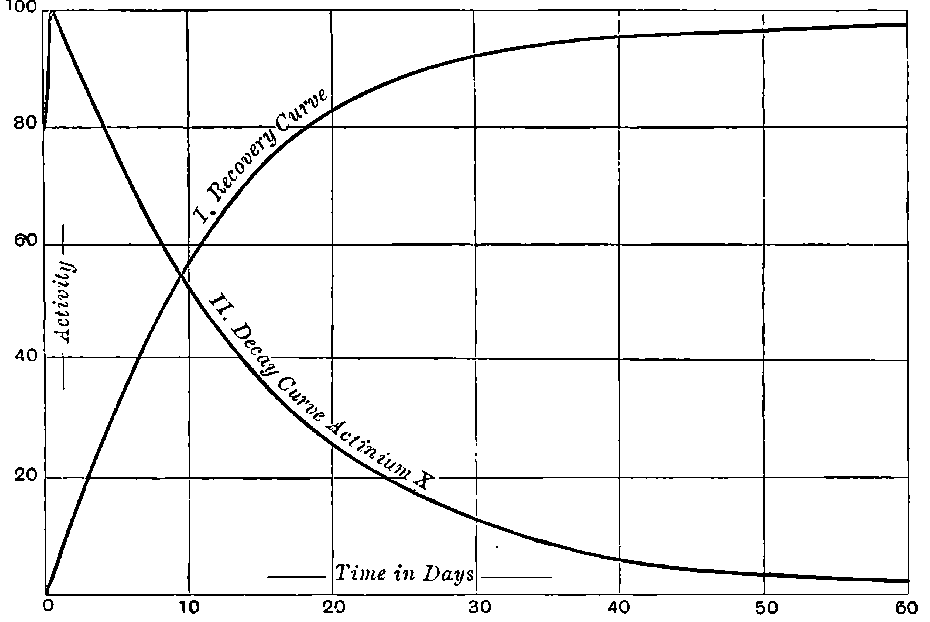
Fig. 82.
The actinium X is produced at a constant rate from the parent matter actinium, and is transformed according to an exponential law with the time. The constant of change λ = ·068 (day)-1, and this value is characteristic of the product actinium X. As in the case of thorium, the above experiments show that the emanation does not arise from actinium itself but from actinium X. The emanation in turn breaks up and gives rise to an active deposit on the surface of bodies.
212. Analysis of the active deposit from the emanation. Debierne[312] observed that the excited activity produced by actinium decayed to half value in about 41 minutes. Miss Brooks[313] showed 367that the curves of decay of the excited activity after removal depended upon the duration of exposure to the emanation. The curves for different times of exposure have already been shown in Fig. 69.
Bronson, using the direct deflection method described in section 69, accurately determined the activity curve corresponding to a short exposure to the actinium emanation. The curve obtained is shown in Fig. 83.
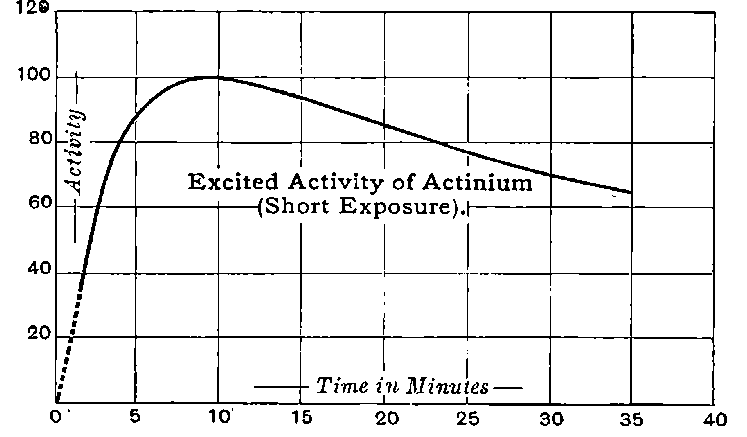
Fig. 83.
This curve is similar in shape to the corresponding curve obtained for the active deposit from thorium, and is explained in a similar way. The activity It at any time t is given by
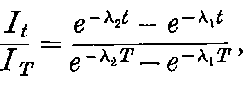
where λ1 and λ2 are two constants, and IT the maximum activity reached after an interval T. After 20 minutes the activity decreased exponentially with the time, falling to half value in 35·7 minutes. This gives the value λ1 = ·0194 (min.)-1. By comparison with the curve, the value of λ2 was found to be ·317 (min.)-1. This corresponds to a change in which half the matter is transformed in 2·15 minutes. Exactly as in the analogous curve for thorium, it can be shown that the matter initially deposited undergoes two changes, the first of which is a rayless one. The same difficulty arises in fixing which of the values of λ refers to 368the first change. An experiment made by Miss Brooks (loc. cit.) shows that the rayless product has the slower period of transformation. The active deposit of actinium was dissolved off a platinum wire and then electrolysed. The anode was found to be active, and the activity fell off exponentially with the time, decreasing to half value in about 1·5 minutes. Allowing for the difficulty of accurately measuring such a rapid rate of decay, this result indicates that the product which gives out rays has the rapid period of 2·15 minutes. The analysis of the active deposit of actinium thus leads to the following conclusions:
(1) The matter initially deposited from the emanation, called actinium A, does not give out rays, and is half transformed in 35·7 minutes.
(2) A changes into B, which is half transformed in 2·15 minutes, and gives out both α and β (and probably γ) rays.
Godlewski found that the active deposit of actinium was very easily volatilized. Heating for several minutes at a temperature of 100° C. was sufficient to drive off most of the active matter. The active deposit is readily soluble in ammonia and in strong acids.
213. Radiations from actinium and its products. Actinium in radio-active equilibrium gives out α, β, and γ rays. Godlewski found several points of distinction between the β and γ rays of actinium and of radium. The β rays of actinium appear to be homogeneous, for the activity measured by an electroscope was found to fall off accurately according to an exponential law with the thickness of matter traversed. The β rays were half absorbed in a thickness of 0·21 mm. of aluminium. This indicates that the β particles are all projected from actinium with the same velocity. In this respect actinium behaves very differently from radium, for the latter gives out β particles whose velocities vary over a wide range.
After the β rays were absorbed, another type of more penetrating rays was observed, which probably corresponds to the γ rays from the other radio-elements. The γ rays of actinium were, however, far less penetrating than those from radium. The activity due to these rays was reduced to one-half after passing 369through 1·9 mms. of lead, while the thickness of lead required in order to absorb half the γ rays of radium is about 9 mms.
The active deposit gave out α and β (and probably γ) rays. It was difficult to decide definitely whether actinium X gave out β as well as α rays. When the actinium X was heated to a red heat, the β activity was temporarily reduced to about half its initial value. This decrease was probably due to the removal of the active deposit, which, we have seen, is readily volatilized by heat. If the β ray activity cannot be further reduced, this would point to the conclusion that actinium X, as well as actinium B, gives out β rays, but the evidence so far obtained is not conclusive.
The ease with which the active deposit is volatilized by heat offers a very simple explanation of the initial peculiarities of the decay and recovery curves (Fig. 82) of actinium X. The activity of actinium X rises at first, but there is no corresponding decrease in the activity of the actinium left behind. It has been shown that the active deposit is soluble in ammonia, and, in consequence, is removed with the actinium X. The products actinium A and B and actinium X, immediately after separation, are in radio-active equilibrium and we should not therefore expect to find any increase of activity after removal, such as is observed in the case of thorium, where thorium A and B are not removed with thorium X. However, in heating the actinium X to drive off the ammonium salts, some of the active deposit is volatilized. After cooling, the amount of the active deposit increases to nearly its old value and there is a corresponding increase of the activity.
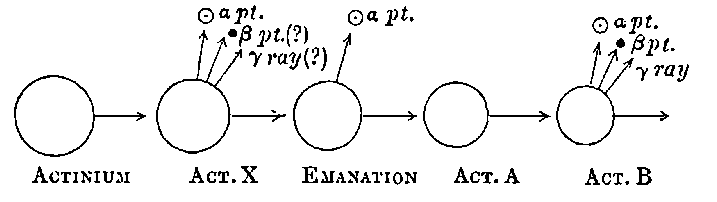
Fig. 84.
214. Products of Actinium. There is one very interesting point of distinction between the radio-active behaviour of thorium and actinium. The latter after removal of actinium X, shows only about 5 per cent. of the original activity, while thorium, after 370removal of Th X, always shows a residual activity of about 25 per cent. of the maximum value. This very small residual activity indicates that actinium, if completely freed from all its products, would not give out rays at all, in other words, the first change in actinium is a rayless one.
The radio-active products of actinium are shown graphically in Fig. 84. Some of their chemical and physical properties are tabulated below.
| Products | Time to be half transformed | Rays | Some Physical and Chemical properties |
|---|---|---|---|
| Actinium | ? | No rays | Insoluble in ammonia |
| Actinium X | 10·2 days | α, (β and γ) | Soluble in ammonia |
| Emanation | 3·9 secs. | α rays | Behaves as a gas |
| Actinium A | 35·7 mins. | No rays | Soluble in ammonia and strong acids. |
| Actinium B | 2·15 mins. | α, β and γ | Volatilized at 100°C. B can be separated from A by electrolysis |
215. Radio-activity of radium. Notwithstanding the enormous difference in their relative activities, the radio-activity of radium presents many close analogies to that of thorium and actinium. Both substances give rise to emanations which in turn produce “excited activity” on bodies in their neighbourhood. Radium, however, does not give rise to any intermediate product between the element itself and the emanation it produces, or in other words there is no product in radium corresponding to Th X in thorium.
Giesel first drew attention to the fact that a radium compound gradually increased in activity after preparation, and only reached a constant value after a month’s interval. If a radium compound is dissolved in water and boiled for some time, or a current of air drawn through the solution, on evaporation it is found that the activity has been diminished. The same result is observed if a solid radium compound is heated in the open air. This loss of activity is due to the removal of the emanation by the process of solution or heating. Consider the case of a radium compound which has been kept for some time in solution in a shallow vessel, exposed to the open air, and then evaporated to dryness. The emanation which, in the state of solution, was removed as fast as it was formed, is now occluded, and, together with the active deposit which it produces, adds its radiations to that of the original radium. The activity will increase to a maximum value when the rate of production of fresh emanation balances the rate of change of that already produced.
372If now the compound is again dissolved or heated, the emanation escapes. Since the active deposit is not volatile and is insoluble in water, it is not removed by the process of solution or heating. Since, however, the parent matter is removed, the activity due to the active deposit will immediately begin to decay, and in the course of a few hours will have almost disappeared. The activity of the radium measured by the α rays is then found to be about 25 per cent. of its original value. This residual activity of radium, consisting entirely of α rays, is non-separable, and has not been further diminished by chemical or physical means. Rutherford and Soddy[314] examined the effect of aspiration for long intervals through a radium chloride solution. After the first few hours the activity was found to be reduced to 25 per cent., and further aspiration for three weeks did not produce any further diminution. The radium was then evaporated to dryness, and the rise of its activity with time determined. The results are shown in the following table. The final activity in the second column is taken as one hundred. In column 3 is given the percentage proportion of the activity recovered.
| Time in days | Activity | Percentage Activity recovered |
|---|---|---|
| 0 | 25·0 | 0 |
| 0·70 | 33·7 | 11·7 |
| 1·77 | 42·7 | 23·7 |
| 4·75 | 68·5 | 58·0 |
| 7·83 | 83·5 | 78·0 |
| 16·0 | 96·0 | 95·0 |
| 21·0 | 100·0 | 100·0 |
The results are shown graphically in Fig. 85.
The decay curve of the radium emanation is shown in the same figure. The curve of recovery of the lost activity of radium is thus analogous to the curves of recovery of uranium and thorium which have been freed from the active products Ur X and Th X respectively. The intensity It of the recovered activity at any time is given by
where I₀ is the final value, and λ is 373the radio-active constant of the emanation. The decay and recovery curves are complementary to one another.
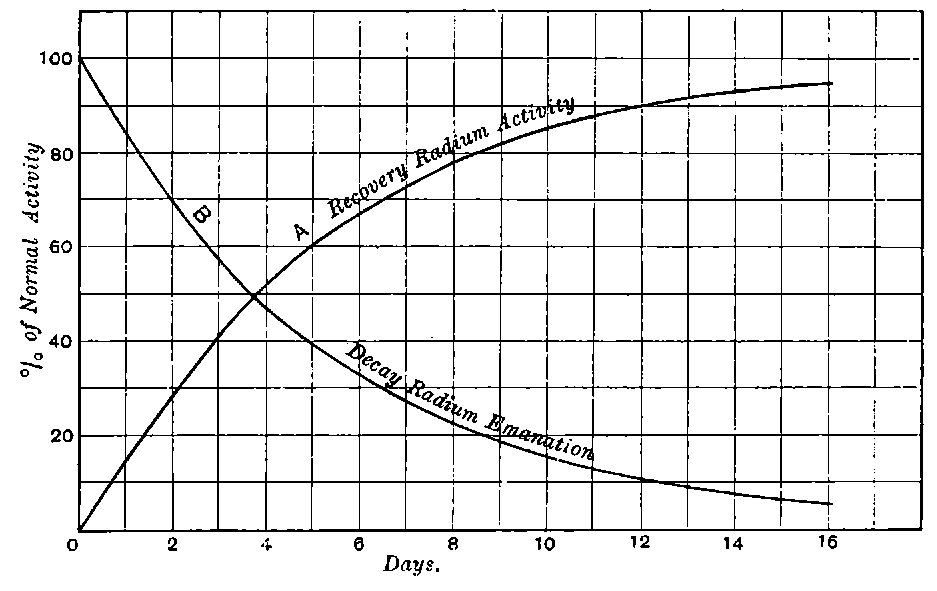
Fig. 85.
Knowing the rate of decay of activity of the radium emanation, the recovery curve of the activity of radium can thus at once be deduced, provided all of the emanation formed is occluded in the radium compound.
When the emanation is removed from a radium compound by solution or heating, the activity measured by the β rays falls almost to zero, but increases in the course of a month to its original value. The curve showing the rise of β and γ rays with time is practically identical with the curve, Fig. 85, showing the recovery of the lost activity of radium measured by the α rays. The explanation of this result lies in the fact that the β and γ rays from radium only arise from the active deposit, and that the non-separable activity of radium gives out only α rays. On removal of the emanation, the activity of the active deposit decays nearly to zero, and in consequence the β and γ rays almost disappear. When the radium is allowed to stand, the emanation begins to accumulate, and produces in turn the active deposit, which gives 374rise to β and γ rays. The amount of β and γ rays (allowing for a period of retardation of a few hours) will then increase at the same rate as the activity of the emanation, which is continuously produced from the radium.
216. Effect of escape of emanation. If the radium allows some of the emanation produced to escape into the air, the curve of recovery will be different from that shown in Fig. 85. For example, suppose that the radium compound allows a constant fraction α of the amount of emanation, present in the compound at any time, to escape per second. If n is the number of emanation particles present in the compound at the time t, the number of emanation particles changing in the time dt is λndt, where λ is the constant of decay of activity of the emanation. If q is the rate of production of emanation particles per second, the increase of the number dn in the time dt is given by
The same equation is obtained when no emanation escapes, with the difference that the constant λ + α is replaced by λ. When a steady state is reached, dn/dt is zero, and the maximum value of n is equal to q/(λ + α).
If no escape takes place, the maximum value of n is equal to q/λ. The escape of emanation will thus lower the amount of activity recovered in the proportion λ/(λ + α). If n₀ is the final number of emanation particles stored up in the compound, the integration of the above equation gives
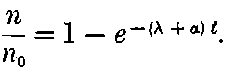
The curve of recovery of activity is thus of the same general form as the curve when no emanation escapes, but the constant λ is replaced by λ + α.
375For example, if α = λ = ¹⁄₄₆₃₀₀₀, the equation of rise of activity is given by
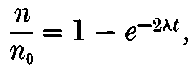
and, in consequence, the increase of activity to the maximum will be far more rapid than in the case of no escape of emanation.
A very slight escape of emanation will thus produce large alterations both in the final maximum and in the curve of recovery of activity.
A number of experiments have been described by Mme Curie in her Thèse présentée à la Faculté des Sciences de Paris on the effect of solution and of heat in diminishing the activity of radium. The results obtained are in general agreement with the above view, that 75 per cent. of the activity of radium is due to the emanation and the excited activity it produces. If the emanation is wholly or partly removed by solution or heating, the activity of the radium is correspondingly diminished, but the activity of the radium compound is spontaneously recovered owing to the production of fresh emanation. A state of radio-active equilibrium is reached, when the rate of production of fresh emanation balances the rate of change in the emanation stored up in the compound. The differences observed in the rate of recovery of radium under different conditions were probably due to variations in the rate of escape of the emanation.
217. It has been shown in section 152 that the emanation is produced at the same rate in the solid as in the solution, and all the results obtained point to the conclusion that the emanation is produced from radium at a constant rate, which is independent of physical conditions. Radium, like thorium, shows a non-separable activity of 25 per cent. of the maximum activity, and consisting entirely of α rays. The β and γ rays arise only from the active deposit. The emanation itself (section 156) gives out only α rays. These results thus admit of the explanation given in the case of thorium (section 136). The radium atoms break up at a constant rate with the emission of α particles. The residue of the radium atom becomes the atom of the emanation. This in turn is unstable and breaks up with the expulsion of an α particle. The emanation is half transformed in four days. We 376have seen that this emanation gives rise to an active deposit. The results obtained up to this stage are shown diagrammatically below.
218. Analysis of the active deposit from radium. We have seen in chapter VIII that the excited activity produced on bodies, by the action of the radium emanation, is due to a thin film of active matter deposited on the surface of bodies. This active deposit is a product of the decomposition of the radium emanation, and is not due to any action of the radiations on the surface of the matter.
The curves showing the variation of the excited activity with time are very complicated, depending not only upon the time of exposure in the presence of the emanation, but also upon the type of radiation used for measurement. The greater portion of the activity of this deposit dies away in the course of 24 hours, but a very small fraction still remains, which then changes very slowly.
It will be shown in this chapter that at least six successive transformations occur in the active deposit. The matter initially produced from the emanation is called radium A, and the succeeding products B, C, D, E, F. The equations expressing the quantity of A, B, C,...... present at any time are very complicated, but the comparison of theory with experiment may be much simplified by temporarily disregarding some unimportant terms: for example, the products A, B, C are transformed at a very rapid rate compared with D. The activity due to D + E + F is, in most cases, negligible compared with that of A or C, being usually less than ¹⁄₁₀₀₀₀₀ of the initial activity observed for A or C. The analysis of the active deposit of radium may thus be conveniently divided into two stages:
(1) Analysis of the deposit of rapid change, which is mainly composed of radium A, B, and C;
(2) Analysis of the deposit of slow change, which is composed of radium D, E, and F.
377219. Analysis of the deposit of rapid change. In the experiments described below, a radium solution was placed in a closed glass vessel. The emanation then collected in the air space above the solution. The rod, to be made active, was introduced through an opening in the stopper and exposed in the presence of the emanation for a definite interval. If the decay was to be measured by the α rays, the rod was made the central electrode in a cylindrical vessel such as is shown in Fig. 18. A saturating voltage was applied, and the current between the cylinders measured by an electrometer. If a very active rod is to be tested, a sensitive galvanometer can be employed, but, in such a case, a large voltage is required to produce saturation. A slow current of dust-free air was continuously circulated through the cylinder, in order to remove any emanation that may have adhered to the rod. For experiments on the β and γ rays, it was found advisable to use an electroscope, such as is shown in Fig. 12, instead of an electrometer. For measurements with the γ rays, the active rod was placed under the electroscope, and before entering the vessel the rays passed through a sheet of metal of sufficient thickness to absorb all the α rays. For measurements with the γ rays, the electroscope was placed on a lead plate 0·6 cms. thick, and the active rod placed under the lead plate. The α and β rays were completely stopped by the lead, and the discharge in the electroscope was then due to the γ rays alone. The electroscope is very advantageous for measurements of this character, and accurate observations can be made simply and readily.
The curve of decay of activity, measured by the α rays, for an exposure of 1 minute in the presence of the radium emanation is shown in Fig. 86, curve BB.
The curve exhibits three stages:—
(1) A rapid decay in the course of 15 minutes to less than 10 per cent. of the value immediately after removal;
(2) A period of 30 minutes in which the activity varies very little;
(3) A gradual decrease almost to zero.
The initial drop decays very approximately according to an 378exponential law with the time, falling to half value in about 3 minutes. Three or four hours after removal the activity again decays according to an exponential law with the time, falling to half value in about 28 minutes. The family of curves obtained for different times of exposure have already been shown in Fig. 67. These results thus indicate:—
(1) An initial change in which half the matter is transformed in 3 minutes;
(2) A final change in which half the matter is transformed in 28 minutes.
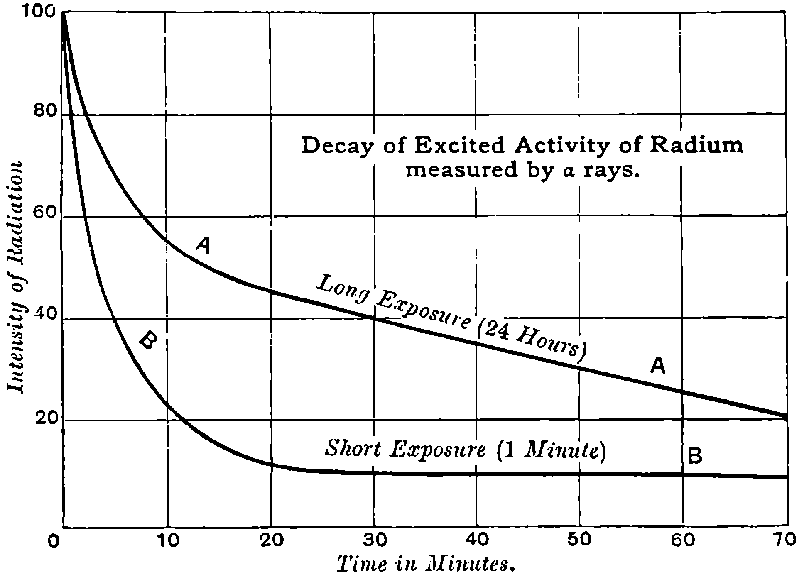
Fig. 86.
Before considering the explanation of the intermediate portion of the curve further experimental results will be considered.
The curve of decay of the excited activity for a long exposure (24 hours) is shown graphically in Fig. 86, curve AA. There is at first a rapid decrease for the first 15 minutes to about 50 per cent. of the initial value, then a slower decay, and, after an interval of about 4 hours, a gradual decay nearly to zero, according to an exponential law with the time, falling to half value in 28 minutes.
379The curves of variation with time of the excited activity when measured by the β rays are shown graphically in Figs. 87 and 88.
Fig. 87 is for a short exposure of 1 minute. Fig. 88 shows the decay for a long exposure of about 24 hours.
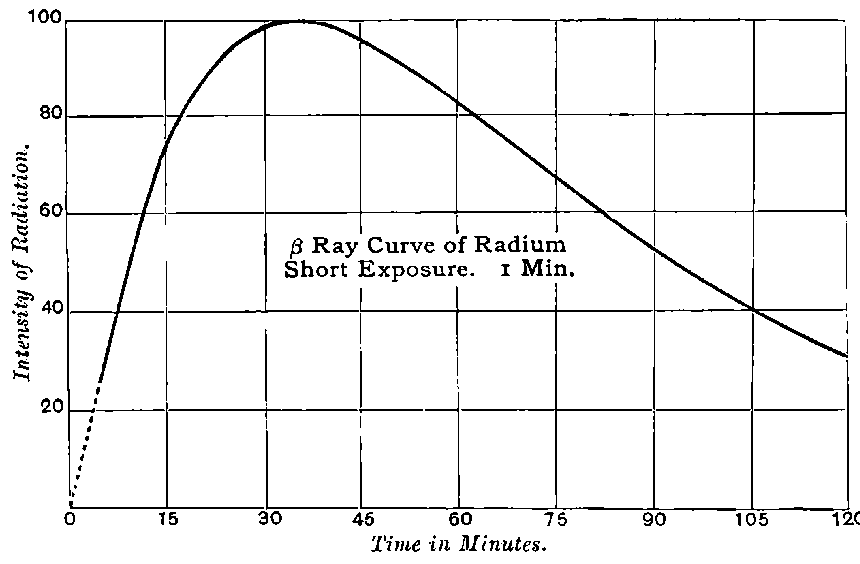
Fig. 87.
The curves obtained for the β rays are quite different from those obtained for the α rays. For a short exposure, the activity measured by the β rays is at first small, then passes through a maximum about 36 minutes after removal. There is then a gradual decrease, and after several hours the activity decays according to an exponential law, falling, as in the other cases, to half value in 28 minutes.
The curve shown in Fig. 88 for the β rays is very similar in shape to the corresponding curve, Fig. 86, curve AA, for the α rays, with the exception that the rapid initial drop observed for the α-ray curve is quite absent. The later portions of the curve are similar in shape, and, disregarding the first 15 minutes after removal, the activity decays at exactly the same rate in both cases.
The curves obtained by means of the γ rays are identical with those obtained for the β rays. This shows that the β and γ rays always occur together and in the same proportion.
380For increase of the time of exposure from 1 minute to 24 hours the curves obtained are intermediate in shape between the two representative limiting curves, Figs. 87 and 88. Some of these curves have already been shown in Fig. 68.
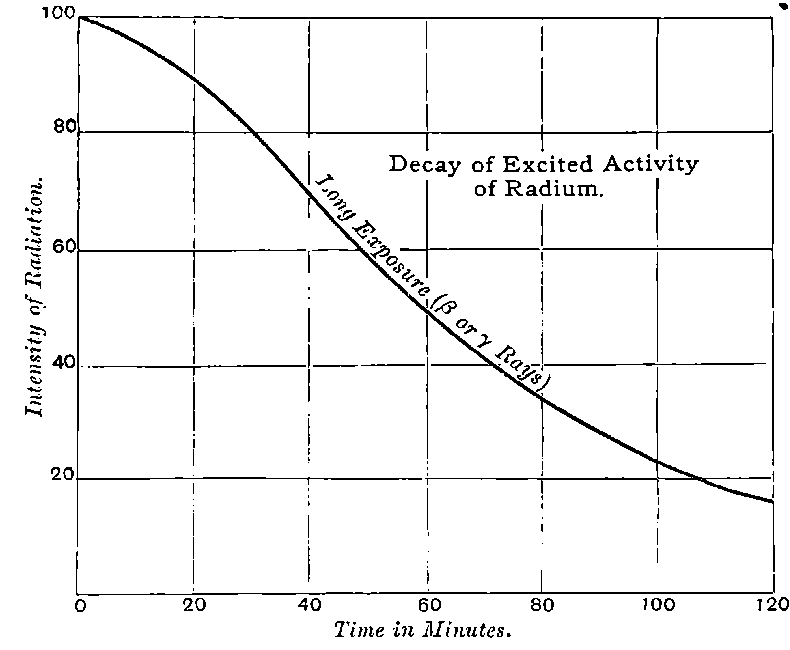
Fig. 88.
220. Explanation of the curves. It has been pointed out that the rapid initial drop for curves A and B, Fig. 86, is due to a change giving rise to α rays, in which half of the matter is transformed in about 3 minutes. The absence of the drop in the corresponding curves, when measured by the β rays, shows that the first 3-minute change does not give rise to β rays; for if it gave rise to β rays, the activity should fall off at the same rate as the corresponding α-ray curve.
It has been shown that the activity several hours after removal decays in all cases according to an exponential law with the time, falling to half value in about 28 minutes. This is the case whether for a short or long exposure, or whether the activity is measured by the α, β, or γ rays. This indicates that the final 28-minute change gives rise to all three types of rays.
381It will be shown that these results can be completely explained on the supposition that three successive changes occur in the deposited matter of the following character[315]:—
(1) A change of the matter A initially deposited in which half is transformed in about 3 minutes. This gives rise only to α rays.
(2) A second “rayless” change in which half the matter B is transformed in 21 minutes.
(3) A third change in which half the matter C is transformed in 28 minutes. This gives rise to α, β, and γ rays.
221. Analysis of the β-ray curves. The analysis of the changes is much simplified by temporarily disregarding the first 3-minute change. In the course of 6 minutes after removal, three quarters of the matter A has been transformed into B and 20 minutes after removal all but 1 per cent. has been transformed. The variation of the amount of matter B or C present at any time agrees more closely with the theory, if the first change is disregarded altogether. A discussion of this important point is given later (section 228).
The explanation of the β-ray curves (see Figs. 87 and 88), obtained for different times of exposure, will be first considered. For a very short exposure, the activity measured by the β rays is small at first, passes through a maximum about 36 minutes later, and then decays steadily with the time.
The curve shown in Fig. 87 is very similar in general shape to the corresponding thorium and actinium curves. It is thus necessary to suppose that the change of the matter B into C does not give rise to β rays, while the change of C into D does. In such a case the activity (measured by the β rays) is proportional to the amount of C present. Disregarding the first rapid change, the activity It at any time t should be given by an equation of the same form (section 207) as for thorium and actinium, viz.,
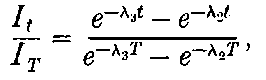
382where IT is the maximum activity observed, which is reached after an interval T. Since the activity finally decays according to an exponential law (half value in 28 minutes), one of the values of λ is equal to 4·13 × 10-4. As in the case of thorium and actinium, the experimental curves do not allow us to settle whether this value of λ is to be given to λ2 or λ3. From other data (see section 226) it will be shown later that it must refer to λ3. Thus λ3 = 4·13 × 10-4 (sec)-1.
The experimental curve agrees very closely with theory if λ2 = 5·38 × 10-4 (sec)-1.
The agreement between theory and experiment is shown by the table given below. The maximum value IT (which is taken as 100) is reached at a time T = 36 minutes.
In order to obtain the β-ray curve, the following procedure was adopted. A layer of thin aluminium was placed inside a glass tube, which was then exhausted. A large quantity of radium emanation was then suddenly introduced by opening a stop-cock communicating with the emanation vessel, which was at atmospheric pressure. The emanation was left in the tube for 1·5 minutes and then was rapidly swept out by a current of air. The aluminium was then removed and was placed under an electroscope, such as is shown in Fig. 12. The α rays from the aluminium were cut off by an interposed screen of aluminium ·1 mm. thick. The time was reckoned from a period of 45 seconds after the introduction of the emanation.
| Time in minutes | Theoretical value of activity | Observed value of activity |
|---|---|---|
| 0 | 0 | 0 |
| 10 | 58·1 | 55 |
| 20 | 88·6 | 86 |
| 30 | 97·3 | 97 |
| 36 | 100 | 100 |
| 40 | 99·8 | 99·5 |
| 50 | 93·4 | 92 |
| 60 | 83·4 | 82 |
| 80 | 63·7 | 61·5 |
| 100 | 44·8 | 42·5 |
| 120 | 30·8 | 29 |
383There is thus a good agreement between the calculated and observed values of the activity measured by the β rays.
The results are satisfactorily explained if it is supposed:—
(1) That the change B into C (half transformed in 21 minutes) does not give rise to β rays;
(2) That the change C into D (half transformed in 28 minutes) gives rise to β rays.
222. These conclusions are very strongly supported by observations of the decay measured by the β rays for a long exposure. The curve of decay is shown in Fig. 88 and Fig. 89, curve I.
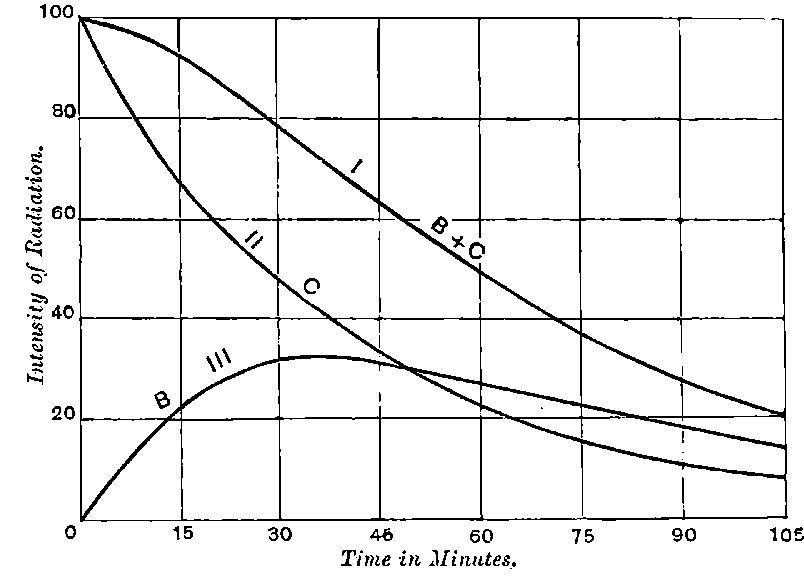
Fig. 89.
P. Curie and Danne made the important observation that the curve of decay C, corresponding to that shown in Fig. 88, for a long exposure, could be accurately expressed by an empirical equation of the form
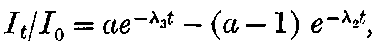
where λ2 = 5·38 × 10-4 (sec)-1 and λ3 = 4·13 × 10-4 (sec)-1, and α = 4·20 is a numerical constant.
I have found that within the limit of experimental error this equation represents the decay of excited activity of radium for a 384long exposure, measured by the β rays. The equation expressing the decay of activity, measured by the α rays, differs considerably from this, especially in the early part of the curve. Several hours after removal the activity decays according to an exponential law with the time, decreasing to half value in 28 minutes. This fixes the value of λ3. The constant α and the value of λ2 are deduced from the experimental curve by trial. Now we have already shown (section 207) that in the case of the active deposit from thorium, where there are two changes of constants λ2 and λ3, in which only the second change gives rise to a radiation, the intensity of the radiation is given by
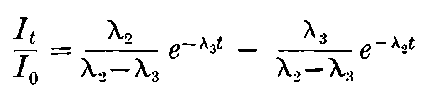
for a long time of exposure (see equation 8, section 198). This is an equation of the same form as that found experimentally by Curie and Danne. On substituting the values λ2, λ3 found by them,
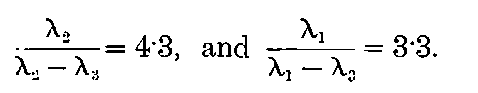
Thus the theoretical equation agrees in form with that deduced from observation, and the values of the numerical constants are also closely concordant. If the first as well as the second change gave rise to a radiation, the equation would be of the same general form, but the value of the numerical constants would be different, the values depending upon the ratio of the ionization in the first and second changes. If, for example, it is supposed that both changes give out β rays in equal amounts, it can readily be calculated that the equation of decay would be
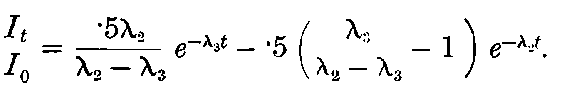
Taking the values of λ2 and λ3 found by Curie, the numerical factor
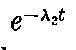
becomes 2·15 instead of 4·3 and 1·15 instead of 3·3. The theoretical curve of decay in this case would be readily distinguishable from the observed curve of decay. The fact that the equation of decay found by Curie and Danne involves the necessity of an initial rayless change can be shown as follows:—
385Curve I (Fig. 89) shows the experimental curve. At the moment of removal of the body from the emanation (disregarding the initial rapid change), the matter must consist of both B and C. Consider the matter which existed in the form C at the moment of removal. It will be transformed according to an exponential law, the activity falling by one-half in 28 minutes. This is shown in curve II. Curve III represents the difference between the ordinates of curves I and II. It will be seen that it is identical in shape with the curve (Fig. 87) showing the variation of the activity for a short exposure, measured by the β rays. It passes through a maximum at the same time (about 36 minutes). The explanation of such a curve is only possible on the assumption that the first change is a rayless one. The ordinates of curve III express the activity added in consequence of the change of the matter B, present after removal, into the matter C. The matter B present gradually changes into C, and this, in its change to D, gives rise to the radiation observed. Since the matter B alone is considered, the variation of activity with time due to its further changes, shown by curve III, should agree with the curve obtained for a short exposure (see Fig. 87), and this, as we have seen, is the case.
The agreement between theory and experiment is shown in the following table. The first column gives the theoretical curve of decay for a long exposure deduced from the equation
taking the value of λ2 = 5·38 × 10-4 and λ3 = 4·13 × 10-4.
| Time in minutes | Calculated values | Observed values |
|---|---|---|
| 0 | 100 | 100 |
| 10 | 96·8 | 97·0 |
| 20 | 89·4 | 88·5 |
| 30 | 78·6 | 77·5 |
| 40 | 69·2 | 67·5 |
| 50 | 59·9 | 57·0 |
| 60 | 49·2 | 48·2 |
| 80 | 34·2 | 33·5 |
| 100 | 22·7 | 22·5 |
| 120 | 14·9 | 14·5 |
386The second column gives the observed activity (measured by means of an electroscope) for a long exposure of 24 hours in the presence of the emanation.
In cases where a steady current of air is drawn over the active body, the observed values are slightly lower than the theoretical. This is probably due to a slight volatility of the product radium B at ordinary temperatures.
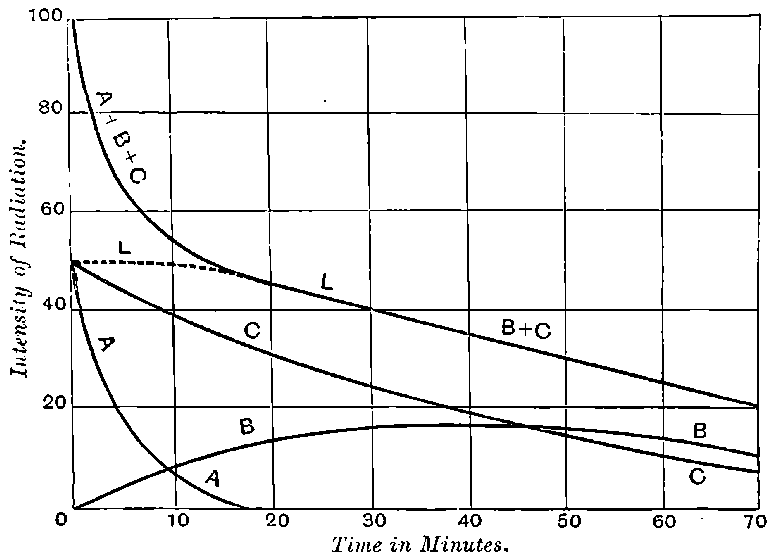
Fig. 90.
223. Analysis of the α-ray curves. The analysis of the decay curves of the excited activity of radium, measured by the α rays, will now be discussed. The following table shows the variation of the intensity of the radiation after a long exposure in the presence of the radium emanation. A platinum plate was made active by exposure for several days in a glass tube containing a large quantity of emanation. The active platinum after removal was placed on the lower of two parallel insulated lead plates, and a saturating electromotive force of 600 volts was applied. The ionization current was sufficiently large to be measured by means of a sensitive high-resistance galvanometer, and readings were taken as quickly as possible after removal of the platinum from the emanation vessel. The initial value of the current (taken 387as 100) was deduced by continuing the curves backwards to meet the vertical axis (see Fig. 90), and was found to be 3 × 10-8 ampere.
| Time in minutes | Current |
|---|---|
| 0 | 100 |
| 2 | 80 |
| 4 | 69·5 |
| 6 | 62·4 |
| 8 | 57·6 |
| 10 | 52·0 |
| 15 | 48·4 |
| 20 | 45·4 |
| 30 | 40·4 |
| 40 | 35·6 |
| 50 | 30·4 |
| 60 | 25·4 |
| 80 | 17·4 |
| 100 | 11·6 |
| 120 | 7·6 |
These results are shown graphically in the upper curve of Fig. 90. The initial rapid decrease is due to the decay of the activity of the matter A. If the slope of the curve is produced backwards from a time 20 minutes after removal, it cuts the vertical axis at about 50. The difference between the ordinates of the curves A + B + C and LL at any time is shown in the curve AA. The curve AA represents the activity at any time supplied by the change in radium A. The curve LL starting from the vertical axis is identical with the curve already considered, representing the decay of activity measured by the β rays for a long exposure (see Fig. 88).
| Time in minutes | Calculated value of activity | Observed value of activity |
|---|---|---|
| 0 | 100 | 100 |
| 10 | 96·8 | 97·0 |
| 20 | 89·4 | 89·2 |
| 30 | 78·6 | 80·8 |
| 40 | 69·2 | 71·2 |
| 50 | 59·9 | 60·8 |
| 60 | 49·2 | 50·1 |
| 80 | 34·2 | 34·8 |
| 100 | 22·7 | 23·2 |
| 120 | 14·9 | 15·2 |
This is shown by the agreement of the numbers in the above table. The first column in the table 388above gives the theoretical values of the activity deduced from the equation
for the values of λ2, λ3 previously employed. The second column gives the observed values of the activity deduced from the decay curve LL.
The close agreement of the curve LL with the theoretical curve deduced on the assumption that there are two changes, the first of which does not emit rays, shows that the change of radium B into C does not emit α rays. In a similar way, as in the curve I, Fig. 89, the curve LL may be analysed into its two components represented by the two curves CC and BB. The curve CC represents the activity supplied by the matter C present at the moment of removal. The curve BB represents the activity resulting from the change of B into C and is identical with the corresponding curve in Fig. 89. Using the same line of reasoning as before, we may thus conclude that the change of B into C is not accompanied by α rays. It has already been shown that it does not give rise to β rays, and the identity of the β and γ-ray curves shows that it does not give rise to γ rays. The change of B into C is thus a “rayless” change, while the change of C into D gives rise to all three kinds of rays.
An analysis of the decay of the excited activity of radium thus shows that three distinct rapid changes occur in the matter deposited, viz.:—
(1) The matter A, derived from the change in the emanation, is half transformed in 3 minutes and is accompanied by α rays alone;
(2) The matter B is half transformed in 21 minutes and gives rise to no ionizing rays;
(3) The matter C is half transformed in 28 minutes and is accompanied by α, β, and γ rays;
(4) A fourth very slow change will be discussed later.
224. Equations representing the activity curves. The equations representing the variation of activity with time are for 389convenience collected below, where λ1 = 3·8 × 10-3, λ2 = 5·38 × 10-4, λ3 = 4·13 × 10-4:—
(1) Short exposure: activity measured by β rays,
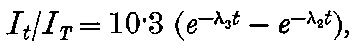
where IT is the maximum value of the activity;
(2) Long exposure: activity measured by β rays,
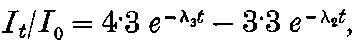
where I₀ is the initial value;
(3) Any time of exposure T: activity measured by the β rays,
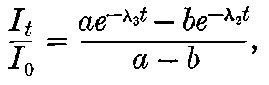
where
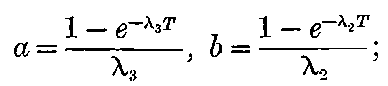
(4) Activity measured by α rays: long time of exposure,
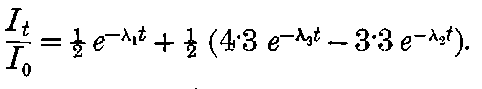
The equations for the α rays for any time of exposure can be readily deduced, but the expressions are somewhat complicated.
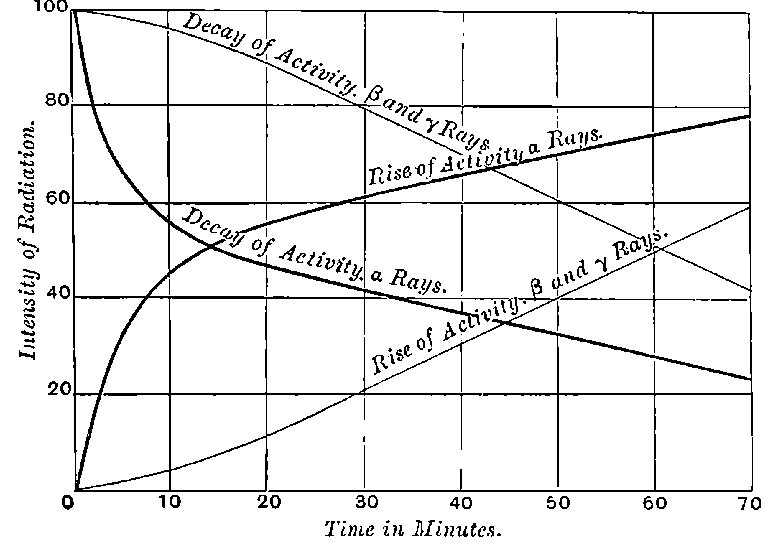
Fig. 91.
225. Equations of rise of excited activity. The curves expressing the gradual increase to a maximum of the excited 390activity produced on a body exposed in the presence of a constant amount of emanation are complementary to the curves of decay for a long exposure. The sum of the ordinates of the rise and decay curves is at any time a constant. This follows necessarily from the theory and can also be deduced simply from à priori considerations. (See section 200.)
The curves of rise and decay of the excited activity for both the α and β rays are shown graphically in Fig. 91. The thick line curves are for the α rays. The difference between the shapes of the decay curves when measured by the α or β rays is clearly brought out in the figure. The equations representing the rise of activity to a maximum are given below.
For the β and γ rays,
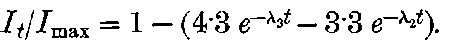
For the α rays,
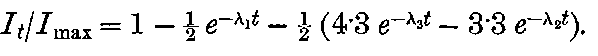
226. Effect of temperature. We have so far not considered the evidence on which the 28-minute rather than the 21-minute change is supposed to take place in the matter C. This evidence has been supplied by some recent important experiments of P. Curie and Danne[316] on the volatilization of the active matter deposited by the emanation. Miss Gates[317] showed that this active matter was volatilized from a platinum wire above a red heat and deposited on the surface of a cold cylinder surrounding the wire. Curie and Danne extended these results by subjecting an active platinum wire for a short time to the action of temperatures varying between 15° C. and 1350° C., and then examining at room temperatures the decay curves not only for the active matter remaining on the wire, but also for the volatilized part. They found that the activity of the distilled part always increased after removal, passed through a maximum, and finally decayed according to an exponential law to half value in 28 minutes. At a temperature of about 630° C. the active matter left behind on the wire decayed at once according to an exponential 391law, falling to half value in 28 minutes. P. Curie and Danne showed that the matter B is much more volatile than C. The former is completely volatilized at about 600° C., while the latter is not completely volatilized even at a temperature of 1300° C. The fact that the matter C, left behind when B is completely volatilized, decays at once to half value in 28 minutes shows that the matter C itself and not B is half transformed in 28 minutes.
Curie and Danne also found that the rate of decay of the active matter varied with the temperature to which the platinum wire had been subjected. At 630° C. the rate of decay was normal, at 1100° C. the activity fell to half value in about 20 minutes, while at 1300° C. it fell to about half value in about 25 minutes.
I have repeated the experiments of Curie and Danne and obtained very similar results. It was thought possible that the measured rate of decay observed after heating might be due to a permanent increase in the rate of volatilization of C at ordinary temperatures. This explanation, however, is not tenable, for it was found that the activity decreased at the same rate whether the activity of the wire was tested in a closed tube or in the open with a current of air passed over it.
These results are of great importance, for they indicate that the rate of change of the product C is not a constant, but is affected by differences of temperature. This is the first case where temperature has been shown to exert an appreciable influence on the rate of change of any radio-active product.
227. Volatility of radium B at ordinary temperature. Miss Brooks[318] has observed that a body, made active by exposure to the radium emanation, possesses the power of exciting secondary activity on the walls of a vessel in which it is placed. This activity was usually about ¹⁄₁₀₀₀ of the whole, but the amount was increased to about ¹⁄₂₀₀ if the active wire was washed in water and dried over a gas flame—the method often adopted to free the wire of any trace of the radium emanation. This effect of producing activity was most marked immediately after removal of the wire from the emanation, and was almost inappreciable ten minutes afterwards.
392The effect was particularly noticeable in some experiments with a copper plate, which was made active by leaving it a short time in a solution of the active deposit from radium. This active solution was obtained by placing an active platinum wire in dilute hydrochloric acid. On placing the copper plate in a testing vessel for a few minutes, and then removing it, activity was observed on the walls of the vessel amounting to about one per cent. of the activity of the copper plate.
It was found that this effect was not due to the emission of an emanation from the active body, but must be ascribed to a slight volatility of radium B at ordinary temperatures. This was proved by observations on the variation of the activity of the matter deposited on the walls of the vessel. The activity was small at first, but rose to a maximum after about 30 minutes, and then decayed with the time. The curve of rise was very similar to that shown in Fig. 87, and shows that the inactive matter radium B was carried to the walls and there changed into C, which gave rise to the radiation observed.
The product B only escapes from the body for a short time after removal. This is a strong indication that its apparent volatility is connected with the presence of the rapidly changing product radium A. Since A breaks up with an expulsion of an α particle, some of the residual atoms constituting radium B may acquire sufficient velocity to escape into the gas, and are then transferred by diffusion to the walls of the vessel.
Miss Brooks observed that the activity was not concentrated on the negative electrode in an electric field but was diffused uniformly over the walls of the vessel. This observation is of importance in considering the explanation of the anomalous effects exhibited by the active deposit of radium, which will be discussed in the following section.
228. Effect of the first rapid change. We have seen that the law of decay of activity, measured by the β or γ rays, can be explained very satisfactorily if the first 3-minute change is disregarded. The full theoretical examination of the question given in sections 197 and 198 and the curves of Figs. 72 and 73 show, however, that the presence of the first change should exercise an 393effect of sufficient magnitude to be detected in measurements of the activity due to the succeeding changes. The question is of great interest, for it involves the important theoretical point whether the substances A and B are produced independently of one another, or whether A is the parent of B. In the latter case, the matter A which is present changes into B, and, in consequence, the amount of B present after A is transformed should be somewhat greater than if B were produced independently. Since the change of A is fairly rapid, the effect should be most marked in the early part of the curve.
In order to examine this point experimentally, the curve of rise of activity, measured by the β rays, was determined immediately after the introduction of a large quantity of the radium emanation into a closed vessel. The curve of decay of activity on a body for a long exposure after removal of the emanation, and the rise of activity after the introduction of the emanation, are in all cases complementary to one another. While, however, it is difficult to measure with certainty whether the activity has fallen in a given time, for example, from 100 to 99 or 98·5, it is easy to be sure whether the corresponding rise of activity in the converse experiment is 1 or 1·5 per cent. of the final amount. Fig. 92, curve I, shows the rise of activity (measured by the β rays) obtained for an interval of 20 minutes after the introduction of the emanation. The ordinates represent the percentage amount of the final activity regained at any time.
Curve III shows the theoretical curve obtained on the assumption that A is a parent of B. This curve is calculated from equation (9) discussed in section 198, and λ1, λ2, λ3 are the values previously found.
Curve II gives the theoretical activity at any time on the assumption that the substances A and B arise independently. This is calculated from an equation of the same form as (8), section 198.
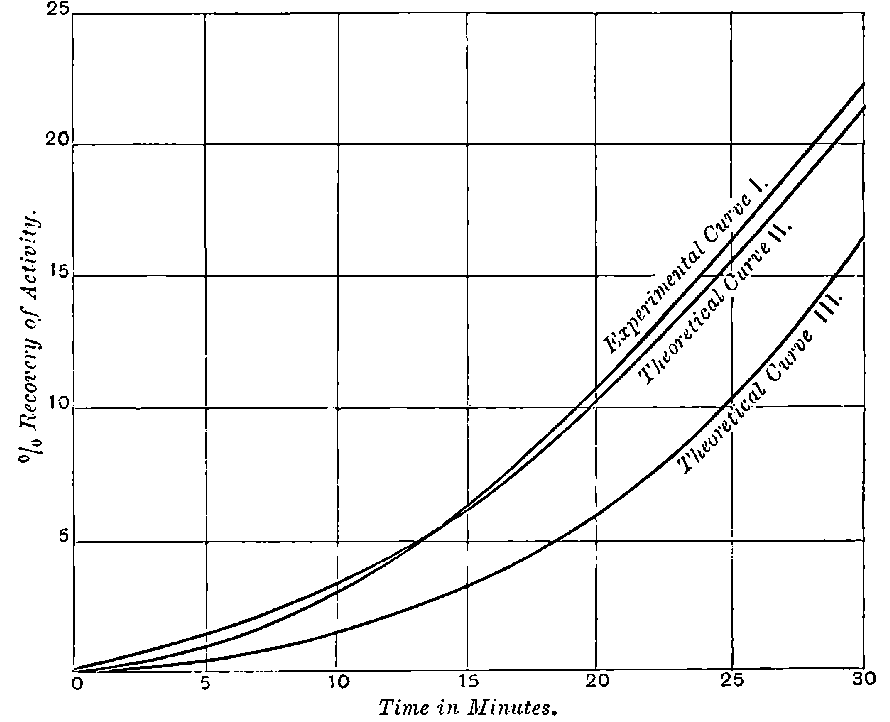
Fig. 92.
It is seen that the experimental results agree best with the view that A and B arise independently. Such a conclusion, however, is of too great importance to be accepted before examining closely whether the theoretical conditions are fulfilled in the experiments. In the first place, it is assumed that the 394carriers which give rise to excited activity are deposited on the surface of the body, to be made active immediately after their formation. There is some evidence, however, that some of these carriers exist for a considerable interval in the gas before their deposit on the body. For example, it is found that if a body is introduced for a short interval, about 1 minute, into a vessel containing the radium emanation, which has remained undisturbed for several hours, the activity after the first rapid decay (see Fig. 86, curve B) is in much greater proportion than if an electric field had been acting for some time previously. This result indicates that the carriers of B and C both collect in the gas and are swept to the electrode when an electric field is applied. I have also observed that if radium emanation, which has stood undisturbed for some time, is swept into a testing vessel, the rise curve is not complementary to the decay curve, but indicates that a large amount of radium B and C was present with the emanation. The experiments of Miss Brooks, previously 395referred to, indicate that radium B does not obtain a charge and so will remain in the gas. Dr. Bronson, working in the laboratory of the writer, has obtained evidence that a large amount of radium D remains in the gas even in a strong electric field. If the matter B exists to some extent in the gas, the difference between the theoretical curves for three successive changes would be explained; for, in transferring the emanation to another vessel, the matter B mixed with it would commence at once to change into C and give rise to a part of the radiation observed.
The equal division of the activity between the products A and C (see Fig. 90) supports the view that C is a product of A, for when radio-active equilibrium is reached, the number of particles of A changing per second is equal to the number of B or C changing per second. If each atom of A and C expels an α particle of the same mass and with the same average velocity, the activity due to the matter A should be equal to that due to the matter C; and this, as we have seen, is the case.
While it is a matter of great difficulty to give a definite experimental proof that radium A and B are consecutive products, I think there is little doubt of its correctness. Accurate determinations of the curves of rise and decay may throw further light on the complicated processes which undoubtedly occur between the breaking up of the atoms of the emanation and the appearance of the active deposit on the electrodes.
229. Relative activity supplied by the α-ray products of radium. There are four products in radium which give out α rays, viz. radium itself, the emanation, radium A and C. If these products are in radio-active equilibrium, the same number of particles of each product are transformed per second and, if each atom breaks up with the emission of one α particle, the number of α particles expelled per second should be the same for each product.
Since, however, the α particles from the different products are not projected with the same velocity, the activity, measured by the ionization current in the usual manner, will not be the same for all products. The activity, when measured by the saturation current between parallel plates at sufficient distance 396apart to absorb all the α rays in the gas, is proportional to the energy of the α particles escaping into the gas.
It has been shown that the minimum activity of radium after removal of the emanation, measured by the α rays, is 25 per cent. of the maximum value. The remaining 75 per cent. is due to the α particles from the other products. Now the activity supplied by radium A and C is nearly the same (section 228). If the emanation is introduced into a cylindrical vessel about 5 cms. in diameter, the activity increases to about twice its initial value owing to the deposit of radium A and C on the surface of the vessel. This shows that the activity of the emanation is of about the same magnitude as that supplied by radium A or C, but an accurate comparison is beset with difficulty, for the emanation is distributed throughout the gas, while radium A and C are deposited on the walls of the vessel. In addition, the relative absorption of the emanation compared with that of radium A and C is not known.
The writer has made some experiments on the decrease of activity of radium immediately after heating to a sufficient temperature to drive off the emanation. The results obtained by this method are complicated by the alteration of the radiating surface in consequence of the heating, but indicate that the emanation supplies about 70 per cent. of the activity of radium A or C.
This points to the conclusion that the α particles from the emanation are projected with less velocity than those from radium C.
The following table shows approximately the activity supplied by the different products of radium in radio-active equilibrium.
| Product | Percentage proportion of total activity |
|---|---|
| Radium | 25 per cent. |
| Emanation | 17 „ |
| Radium A | 29 „ |
| Radium B | 0 „ |
| Radium C | 29 „ |
The products of radium and their radiation are graphically shown later in Fig. 95.
397230. Active deposit of radium of slow transformation. It has been pointed out (section 183) that a body, exposed in the presence of the radium emanation, does not lose all its activity for a long time after removal; a small residual activity is always observed. The magnitude of this residual activity is dependent not only upon the amount of emanation employed, but also upon the time of exposure of the body in the presence of the emanation. For an exposure of several hours in the presence of the emanation, the residual activity is less than one-millionth of the activity immediately after removal.
An account will now be given of some investigations made by the writer[319] on the nature of this residual activity and the chemical properties of the active matter itself. It is first of all necessary to show that the residual activity arises in consequence of a deposit of radio-active matter, and is not due to some action of the intense radiations to which the body made active has been subjected.
The inside of a long glass tube was covered with equal areas of thin metal, including aluminium, iron, copper, silver, lead, and platinum. A large amount of radium emanation was introduced into the tube, and the tube closed. After seven days the metal plates were removed, and, after allowing two days to elapse for the ordinary excited activity to disappear, the residual activity of the plates was tested by an electrometer. The activity of the plates was found to be unequal, being greatest for copper and silver, and least for aluminium. The activity of copper was twice as great as that of aluminium. After standing for another week the activity of the plates was again tested. The activity of each had diminished in the interval to some extent, but the initial differences observed had to a large extent disappeared. After reaching a minimum value the activity of each plate slowly but steadily increased at the same rate. After a month’s interval the activity of each of the plates was nearly the same, and more than three times the minimum value. The initial irregularities in the decay curves of the different metals are, in all probability, due to slight but different degrees of absorption of the radium emanation by the metal plates, the absorption being greatest for copper and silver and least for 398aluminium. As the occluded emanation was slowly released or lost its activity, the activity of the metal fell to a limiting value. The absorption of the radium emanation by lead, paraffin, and caoutchouc has been noticed by Curie and Danne (section 182).
The residual activity on the plates comprised both α and β rays, the latter being present, in all cases, in a very unusual proportion. The equality of the activity and the identity of the radiation emitted from each plate show that the residual activity is due to changes of some form of matter deposited on the plates, and that it cannot be ascribed to an action of the intense radiations; for if such were the case, it would be expected that the activity produced on the different plates would vary not only in quantity, but also in quality. This result is confirmed by the observation that the active matter can be removed from a platinum plate by solution in sulphuric acid, and has other distinctive chemical and physical properties.
The variation with time of the residual activity measured by the α rays will first be considered. A platinum plate was exposed in the presence of the radium emanation for seven days. The amount of emanation initially present was equal to that obtained from about 3 milligrams of pure radium bromide. The plate immediately after removal gave a saturation-current, measured between parallel plates by a galvanometer, of 1·5 × 10-7 ampere. Some hours after removal, the activity decayed according to an exponential law with the time, falling to half value in 28 minutes. Three days after removal the active plate gave a saturation-current, measured by an electrometer, of 5 × 10-13 ampere; i.e. ¹⁄₃₀0,000 of the initial activity. The activity was observed to increase steadily with the time. The results are shown in Fig. 93, where the time is reckoned from the middle of the time of exposure to the emanation.
The curve is initially nearly a straight line passing through the origin. The activity increases with the time for the interval of eight months over which the observations have extended. The latter portions of the curve, however, fall below the tangent to the curve drawn through the origin, showing that the activity is not increasing proportionately with the time.
The active deposit, obtained in a different manner, has been 399examined for a still longer period. The emanation from 30 milligrams of radium bromide was condensed in a glass tube and then sealed. After a month’s interval, the tube was opened and dilute sulphuric acid introduced. The acid dissolved off the active deposit in the tube and on driving off the acid by heat, a radio-active residue was obtained. The activity of this residue, measured by the α rays, steadily increased for a period of 18 months, but the curve of variation of activity with time plotted as in Fig. 93 tends to become more flattened, and is obviously approaching a maximum value.
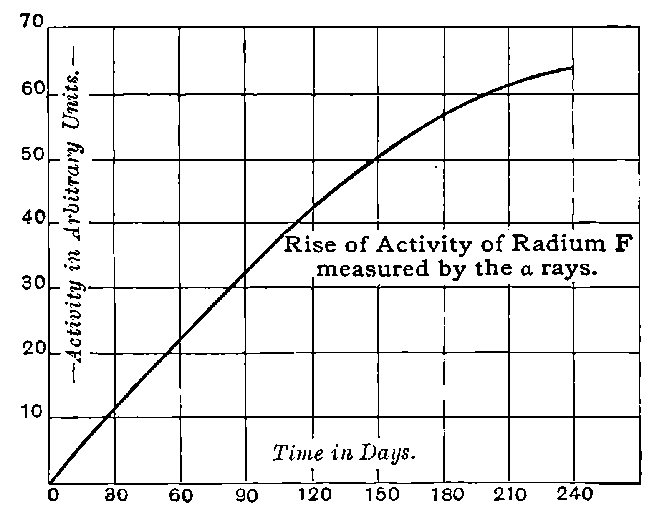
Fig. 93.
The explanation of this curve will be considered later in section 236.
231. Variation of the β ray activity. The residual activity consists of both α and β rays, the latter being present initially in an unusually large proportion. The proportion of α to β rays from the platinum plate, one month after removal, was at the most one-fiftieth of that from a thin film of radium bromide in radio-active equilibrium. Unlike the α ray activity, the activity measured by the β rays remains constant after the active deposit is about one month old, and, in consequence, the proportion of α to β rays steadily increases with the time. The experiments 400showed that the intensity of the β rays did not vary much, if at all, over a further period of eighteen months. The want of proportionality between the α and β rays shows that the two types of rays arise from different products. This conclusion is confirmed by experiments, to be described later, which show that the products giving rise to α and β rays can be temporarily separated from one another by physical and chemical means.
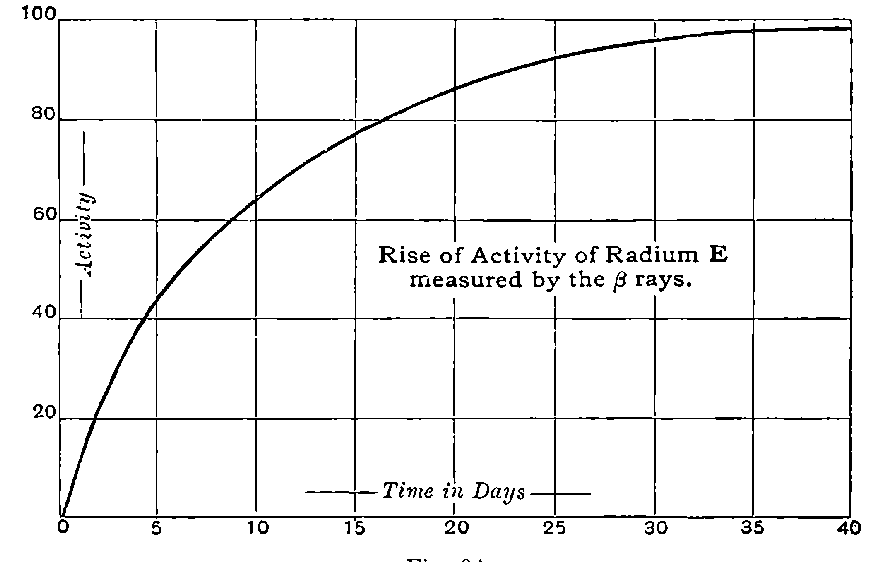
Fig. 94.
If observations of the active deposit are begun shortly after its formation, it is found that the activity, measured by the β rays, is small at first, but increases with the time, reaching a practical maximum about 40 days later. Experiments were made on a platinum plate, which was exposed for 3·75 days in a vessel containing the radium emanation. The observations of the β ray activity began 24 hours after removal. The results are shown in Fig. 94, where the time was measured from the middle of the time of exposure to the emanation. Similar results were obtained for a negatively charged wire exposed to the emanation. The curve, if produced back to the origin, is seen to be very similar to the recovery curves of Ur X, and other active products, and can be expressed by the equation
where I₀ is the maximum 401activity. The activity reaches half its final value in about six days, and the value of λ is equal to ·115 (day)-1. We have shown in section 203 that a rising curve of this character indicates that the β ray activity arises from a product which is supplied at a constant rate from a primary source. Before discussing in detail the explanation of these curves, showing the rise with time of the α and β ray activity, further experimental results will be considered.
232. Effect of temperature on the activity. A platinum plate, made active in the manner described, was exposed to varying temperatures in an electric furnace, and the activity tested at atmospheric temperature after exposure. Four minutes’ exposure in the furnace, at first at 430° C., and afterwards at 800° C., had little, if any, effect on the activity. After four minutes at about 1000° C. the activity decreased about 20 per cent., and a further exposure of eight minutes at a temperature of about 1050° C. almost completely removed the α ray activity. On the other hand, the β ray activity, when measured immediately after removal, was not altered by the heating, but exposure to a still higher temperature caused it to decrease. These results show that the active matter consists of two kinds. The part which emits β rays is not volatile at 1000° C., but the other part, which emits α rays, is almost completely volatilized at that temperature.
It was found, however, that the β ray activity after heating to about 1000° was not permanent, but decayed according to an exponential law with the time, the activity decreasing to half value in about 4·5 days. From the recovery curve of the β ray activity already considered, it was to be expected that the activity would decay to half value in six days. This difference in the periods is possibly due to an effect of the high temperature in altering the rate of decay of radium E. The period of six days is more probably correct. The results obtained on the rise and decay of the β rays, taken together, show:—
(1) That the product giving β rays is supplied at a constant rate from some parent matter of very slow rate of change.
(2) That this parent matter is volatilized at or below 1000° C., and the β ray product is left behind. Since the parent 402matter is removed, the product immediately begins to lose its activity at its characteristic rate, viz. the activity falls to half value in about six days.
233. Separation of the constituents by means of a bismuth plate. The active matter of slow decay was obtained in solution by introducing dilute sulphuric acid into a glass tube in which the emanation from 30 milligrams of radium bromide had been stored for a month. The solution showed strong activity and gave out both α and β rays, the latter, as in other cases, being present in an unusually large proportion.
When a polished bismuth disk was kept for some hours in the solution, it became strongly active. The active matter deposited on the bismuth gave out α rays, but no trace of β rays. After several bismuth disks had been successively left in the solution, the active matter, which emits α rays, was almost completely removed. This was shown by evaporating down the solution after treatment. The β ray activity remained unchanged, but that of the α rays had been reduced to about 10 per cent. of its original value. Three bismuth disks, made active in this way, were set aside and their activity measured at regular intervals. The activity fell off according to an exponential law with the time during the 200 days since their removal, while that of each fell to half value on an average in about 143 days.
At the same time it was observed that the solution, from which the α ray activity was removed, gradually regained its activity, showing that the active substance which gave out α rays was continuously produced from the matter left behind in the solution.
234. Explanation of the results. We have seen that a close examination of the active deposit of slow change has disclosed,
(1) the presence of a β ray product which loses half of its activity in about six days;
(2) the presence of an α ray product, which is deposited on bismuth and is volatilized at 1000° C. This product loses half of its activity in 143 days;
(3) the presence of a parent substance, which produces the β ray product at a constant rate.
403This parent product must be transformed very slowly since the β ray product, which arises from it, soon reaches an equilibrium value, which does not change appreciably over a period of more than one year. The experimental evidence points to the conclusion that the parent product does not give rise to β rays, but that the β rays arise entirely from the next product. This parent product cannot give rise to α rays, for we have seen that the initial α ray activity is at first extremely small, but increases steadily with the time for a period of at least eighteen months. Thus the parent product does not give rise to either α or β rays, and must be a “rayless” product.
The first three transition products of the radium emanation, viz. radium A, B and C, have already been analysed, and shown to be consecutive. It thus seems probable that the active deposit of slow change must arise from the successive transformations of the last product radium C. The results already obtained can be completely explained if it is supposed that three transition products, viz. radium D, E and F, are present in the active deposit of slow rate of change. The properties of these products are summarized below.
Radium D is a rayless product of very slow rate of change. It will be shown later that it is half transformed in about 40 years. It is volatile below 1000° C. and is soluble in strong acids.
Radium E is produced from radium D. In breaking up, it emits β (and probably γ) rays but no α rays. It is half transformed in about 6 days and is not so volatile as radium D and F.
Radium F is produced from radium E. It emits only α rays and is half transformed in 143 days. This substance in solution attaches itself to bismuth. It is volatile at about 1000° C.
Apart from their value and interest in showing the stages of transformation of the radium atom, the results of this analysis have an important bearing upon the origin of some of the well-known radio-active substances separated from pitchblende; for it will be shown later that the product radium F is the radio-active substance present in radio-tellurium and probably also in polonium. 404In addition, there is very strong evidence that the radio-active lead obtained by Hofmann contains the three products radium D, E and F together.
The changes of radium as far as they are at present known, are shown diagrammatically in Fig. 95. It is possible that further investigation will show that the transformation does not end with radium F.
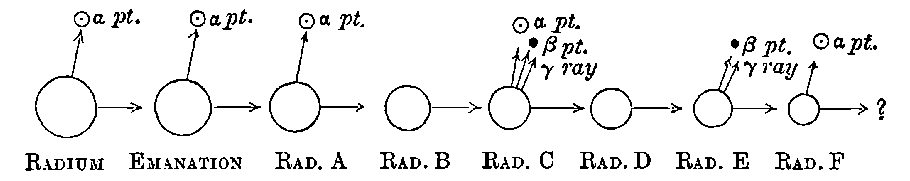
Fig. 95.
While we have shown that radium D is the parent of E, we have not given any conclusive evidence that E is the parent of F. This evidence is, however, supplied by the following experiment. A platinum plate, made active in the manner already described, was placed in an electric furnace and heated for four minutes at about 1000° C. Most of the products D and F were volatilized, but E was left behind. Since the parent matter D was removed, E at once commenced to lose its β ray activity. At the same time it was observed that the small α ray activity, left behind on the platinum plate, increased rapidly at first and then more slowly, as the activity of E became smaller and smaller. This experiment shows conclusively that E was the parent of F, the α ray product.
235. Rate of transformation of radium D. It has been observed experimentally that each of the products of radium, which emit α rays, supplies about an equal proportion of the activity of radium when in radio-active equilibrium. Since, when equilibrium is reached, the same number of particles of each of the successive products must break up per second, this is an expression of the fact that every atom of each product breaks up with the expulsion of an equal number (probably one) of α particles. Now radium D is directly derived from radium C, and, since the rate of change of D is very slow compared with that of C, the number of particles of D initially present must be very nearly equal to the number of particles of radium C which break up 405during the time that radium D is being formed. Now D does not itself give out rays, but the succeeding product E does. The products D and E are practically in radio-active equilibrium one month after D is set aside, and the variation of the β ray activity of E then serves as a measure of the variation of the parent product D. Suppose that a vessel is filled with a large quantity of radium emanation. After several hours, the product radium C, which emits β rays, reaches a maximum value, and then decreases at the same rate as the emanation loses its activity, i.e. it falls to half value in 3·8 days. If N1 is the number of β particles expelled from radium C at its maximum value, the total number Q1 of β particles expelled during the life of the emanation is given approximately by
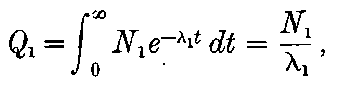
where λ1 is the constant of change of the emanation.
After the emanation has disappeared, and the final products D + E are in radio-active equilibrium, suppose that the number of β particles N2 expelled per second by radium E is determined. If Q2 is the total number of particles expelled during the life of D + E, then Q2 as before is approximately given by Q2 = N2/λ2 where λ2 is the constant of change of radium D. Now we have seen that if each particle of C and of E gives rise to one β particle, it is to be expected that
The ratio N2/N1 was determined by measuring the activity due to the β rays from C and E in the same testing-vessel. Then, since N2/N1 is known, and also the value of λ1, the value of the constant of change, λ2, of radium D is obtained. In this way it was calculated that D is half transformed in about 40 years.
In the above calculations it is assumed, as a first approximation, that the β rays from C and E have the same average velocity. This is probably not accurately the case, but the above number certainly serves to fix the order of magnitude of the period of the 406product D. This calculation is confirmed by observations to be given later on the amount of D and E in old radium.
It may be of interest to mention that the writer calculated the period of radium F by a similar method, before its value was experimentally determined, and found that F should be half transformed in about one year. This is not very different from the experimental value of 143 days found later. In addition, it was assumed in the calculation that the α particles from C and F were projected with the same velocity, and in consequence produced the same amount of ionization. In practice, however, it is found that the α particle of F is absorbed in about half the distance of the α particles of C, and in consequence produces only about half of the ionization of the latter. If this correction were made, the calculated period for half transformation would be six months instead of one year.
A table of the transformation products of radium, together with some of their physical and chemical properties, is given below.
| Transformation Products | Time to be half transformed | Rays | Chemical and Physical Properties |
|---|---|---|---|
| Radium | 1200 years | α rays | — |
| Emanation | 3·8 days | α rays | Chemically inert gas; condenses at -150° C. |
| Radium A (active deposit of rapid change) | 3 mins. | α rays | Behaves as solid; deposited on the surface of bodies; concentrated on cathode in electric field. Soluble in strong acids; volatile at a white heat. B is more volatile than A or C. |
| :: B (same) | 21 mins. | no rays | Same |
| :: C (same) | 28 mins. | α, β, γ rays | Same |
| :: D (active deposit of slow change) | about 40 years | no rays | Soluble in strong acids and volatized below 1000° C. |
| :: E (same) | 6 days | β (and γ) | Non-volatile at 1000°C. |
| :: F (same) | 143 days | α rays | Volatile at 1000° C; deposited from solution on to bismuth plate. |
| ? | — | — | — |
407 236. Variation of the activity over long periods of time. We are now in a position to calculate the variation of the α and β ray activity of the active deposit over long periods of time. If it is supposed that the matter initially deposited consists only of D, the amounts P, Q and R of radium D, E and F existing at any later time are given by the equations 3, 4, 5, section 197.
Since, however, the intermediate product E has a much more rapid rate of change than D or F, the equations can be simplified, without much loss of accuracy, by disregarding the change E, and by supposing that D gives out β rays and changes directly into the α ray product F.
Let λ1, λ2 be the constants of change D and F respectively. Let n₀ be the number of particles of D present initially. Then using the notation of section 197, the amount P of radium D at any time t is given by
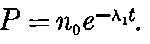
The amount Q of radium F is given by
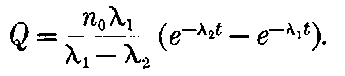
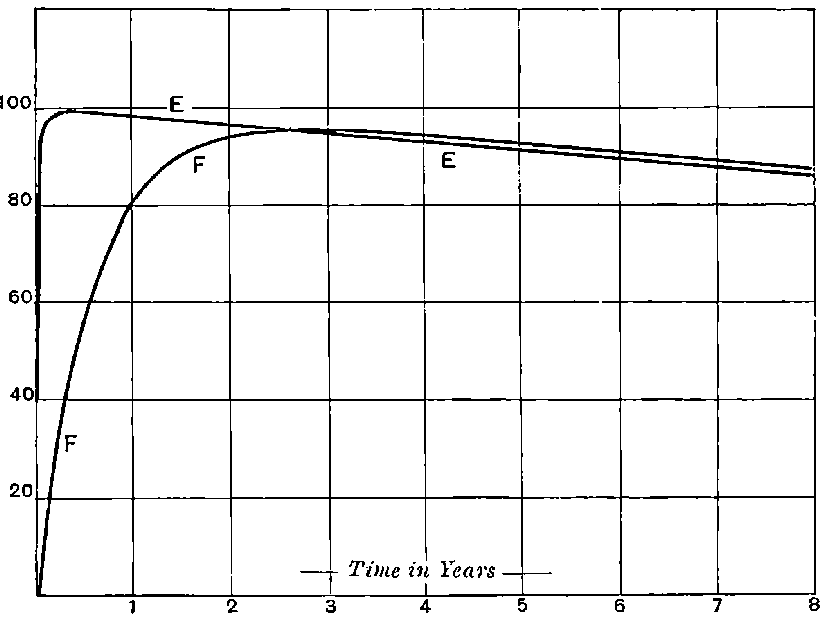
Fig. 96.
The number of β particles emitted by D + E per second, some 408months afterwards, is
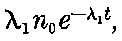
and the number of α particles emitted by radium F is
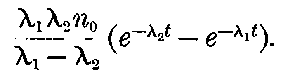
The results are shown graphically in Fig. 96, by the curves EE and FF, in which the ordinates represent the number of β and α particles expelled per second by the products D and F respectively. The complete calculation for three changes shows that the number of β particles soon reaches a practical maximum, and then decays nearly exponentially with the time, falling to half value in 40 years. The number of α particles expelled per second increases for several years, but reaches a maximum after 2·6 years and then diminishes, finally falling off exponentially with the time to half value in 40 years.
The experimental curve of the rise of α ray activity, shown in Fig. 93, as far as it has been determined, lies accurately on this curve, if the maximum is calculated from the above theory. The observed activity after a period of 250 days is marked by the point X on the curve.
237. Experiments with old radium. Since the substance radium D is produced from radium at a constant rate, the amount present mixed with the radium will increase with its age. The writer had in his possession a small quantity of impure radium chloride, kindly presented by Professors Elster and Geitel four years before. The amount of radium D present in it was tested in the following way:—The substance was dissolved in water and kept continuously boiling for a period of about six hours. Under these conditions the emanation is removed as rapidly as it is formed, and the β rays from the radium, due to the product radium C, practically disappear. A newly prepared specimen of radium bromide under these conditions retains only a fraction of 1 per cent. of its original β radiation. The old radium, however, showed (immediately after this treatment) an activity measured by the β rays of about 8 per cent. of its original amount. The activity could not be reduced any lower by further boiling or aspiration of air through the solution. This residual β ray activity was due to the product radium E stored up in the radium. The 409β ray activity due to radium E was thus about 9 per cent. of that due to radium C. Disregarding the differences in the absorption of the β rays, when the activity of the product E in radium reaches a maximum value, the β ray activity due to it should be the same as that due to C. Since the parent product D is half transformed in forty years, the amount present in the radium after four years should be about 7 per cent. of the maximum amount; i.e. it should show a β ray activity of about 7 per cent. of that due to radium C. The observed and calculated values (7 and 9 per cent. respectively) are thus of the same order of magnitude. The amount of β rays from radium E present in pure radium bromide about one year old was about 2 per cent. of the total.
The amount of radium F present in old radium was measured by observations of the activity imparted to a bismuth disk left for several days in the solution, and was found to be of the same order as the theoretical value. Radium F is not deposited to an appreciable extent on the bismuth from a water solution of radium bromide. If, however, a trace of sulphuric acid is added to the solution, the radium F is readily deposited on the bismuth. The addition of sulphuric acid to the radium solution practically effected a separation of radium D, E and F from the radium proper; for the latter was precipitated as sulphate and the products D, E and F remained in solution. After filtering, the solution contained the greater proportion of the products D, E, and F and very little radium.
238. Variation of the activity of radium with time. It has been shown that the activity of freshly prepared radium increases at first with the time and practically reaches a maximum value after an interval of about one month. The results already considered show that there is a still further slow increase of activity with the time. This is the case whether the activity is measured by the α or β rays. It will be shown later that radium is probably half transformed in about 1000 years. From this it can readily be calculated that after a lapse of about 200 years the amount of the products radium D, E and F will have reached a maximum value. The same number of atoms of each of the products C and E will then break up per second. If each atom of these products in disintegrating throws off an equal number 410(probably one) of β particles, the number of β particles thrown off per second will be twice as great as from radium a few months old. The number will increase at first at the rate of about 2 per cent. a year.
Similar considerations apply to the α ray activity. Since, however, there are four other products of radium besides radium itself which expel α particles, the number of α particles emitted per second from old radium will not be more than 25 per cent. greater than the number from radium a few months old. The activity measured by the α rays will thus not increase more than 25 per cent., and probably still less, as the α particles from radium F produce less ionization than the α particles expelled from the other radium products. The activity of radium will consequently rise to a maximum after 200 years and then slowly die away with the time.
239. Presence of these products in pitchblende. The products radium D, E and F must be present in pitchblende in amounts proportional to the quantity of radium present, and should be capable of separation from the mineral by suitable chemical methods. The radio-active properties of these substances, if obtained in the pure state, are summarized below.
Radium D when first separated, should give out very little α or β radiation. The β ray activity will rapidly increase, reaching half its maximum value in 6 days. The α ray activity will at first increase nearly proportionately with the time, and will reach a maximum value after an interval of about 3 years. The α and β ray activity, after reaching a maximum, will finally decay, the activity falling to half value in about 40 years. Since radium D is half transformed in 40 years, and radium in 1200 years, the maximum β ray activity of radium D, weight for weight, will be about 300 times that of radium.
The α ray activity, at any time, will be removed by placing a bismuth disk in the solution.
Radium F, after separation, will give out only α rays. Its activity, after separation, will decrease according to an exponential law, falling to half value in 143 days. Since radium in radio-active equilibrium contains four products which emit α rays, the number of α particles expelled per second from radium F will, weight for weight, be about 800 times as numerous as from new radium in 411radio-active equilibrium. Since the α particles from radium F produce only about half as much ionization as the α particles from the other radium products, the activity of radium F, measured by the electric method, will be about 400 times that of radium.
240. Origin of radio-tellurium and polonium. It is now necessary to consider whether these products of radium have been previously separated from pitchblende, and known by other names.
We shall first consider the α ray product, radium F. The radio-tellurium of Marckwald and the polonium of Mme Curie both resemble radium F in giving out only α rays, and in being deposited on a bismuth disk from a solution. If the active constituent present in radio-tellurium is the same as radium F, its activity should decay at the same rate as the latter. The writer[320] has carefully compared the rates of decay of the activity of radium F and of the radio-tellurium of Marckwald and found them to be the same within the limits of experimental error. Both lose half of their activity in about 143 days[321]. A similar value of the rate of decay of radio-tellurium has been obtained by Meyer and Schweidler[322].
The experiments on radio-tellurium were made upon the active bismuth plates supplied by Dr Sthamer of Hamburg, which were prepared under Marckwald’s directions.
An additional proof[323] of the identity of these two products was obtained by comparing the absorption of the α rays by aluminium foil. The α rays from different products are projected with different velocities, and, in consequence, are unequally absorbed by matter. The absorption of the rays from the two products by aluminium foil agreed very closely, indicating the probable identity of the substances from which they were emitted.
There can thus be no doubt that the active constituent present in the radio-tellurium of Marckwald is identical with the product radium F. This is a very interesting result, and shows how the close examination of the successive transformations of the radio-active bodies may throw light on the origin of the various substances found in pitchblende.
412We have already seen (section 21) that Marckwald, by special chemical methods, was able to obtain a few milligrams of very active substance by working over 2 tons of pitchblende. We have already seen (section 239) that this substance, if obtained in the pure state, should be about 400 times as active as radium. Comparative measurements of the activity of this substance with radium will thus indicate the amount of impurity that is present with the former. This method should be of value in purifying radium F for the purpose of determining its spectrum, which has not yet been observed.
241. Polonium. Since the separation of the active substance by Marckwald, called by him radio-tellurium, there has been some discussion as to whether the active constituent is the same as that present in the polonium of Mme Curie. Both of these substances have similar radio-active and chemical properties, but the main objection to the view that the active constituents were identical has rested on an early statement of Marckwald that the activity of one of his very active preparations did not decay appreciably in the course of six months. This objection is now removed, for we have seen that the activity of radio-tellurium does decay fairly rapidly. It was early recognised that the activity of the polonium, separated from pitchblende by the methods of Mme Curie, was not permanent, but decayed with the time. Observations on the rate of decay have not been very precise, but Mme Curie states that some of her preparations lost half of their activity in about six months but in others the rate of decay was somewhat smaller. It is possible that the initial differences observed in the rates of decay of different specimens of polonium may be due to the presence of some radium D with the polonium. The polonium in my possession lost its activity fairly rapidly, and was reduced to a small portion of its value in the course of about four years. Rough observations of its activity, made from time to time, showed that its activity diminished to half value in about six months. If it is identical with radio-tellurium, the activity should decay to half value in 143 days, and I think there is little doubt that more accurate measurement will prove this to be the case.
While the proof of the identity of the active constituent in polonium is not so definite as for radio-tellurium, I think there can 413be no reasonable doubt that these substances both contain the same active substance, which is the seventh transformation product of radium. Marckwald has noticed some chemical differences in the behaviour of polonium and radio-tellurium, but little weight can be attached to such observations, for it must be remembered that the active constituent in both cases is present in minute quantity in the material under examination, and that the apparent chemical properties of the active substance are much influenced by the presence of impurities. The most important and trustworthy test rests upon the identity of the radiations and the period of decay.
241 A. Origin of radio-active lead. Some experiments will now be discussed which show that the radio-lead first separated from pitchblende by Hofmann (section 22) contains the products radium D, E and F. Hofmann has observed that the activity of this substance did not appreciably decay in the course of several years. In some recent experiments, Hofmann, Gonder and Wölfl[324] have made a close chemical examination of the radio-active lead, and have shown the presence of two radio-active constituents, which are probably identical with the products radium E and F. The radio-active measurements were unfortunately not very precise, and the periods of change of the separated products have not been examined very closely.
Experiments were made on the effect of adding substances to a solution of radio-lead, and then removing them by precipitation. Small quantities of iridium, rhodium, palladium, and platinum, in the form of chlorides, were left in the solution for three weeks, and then precipitated by formalin or hydroxylamine. All of these substances were found to give out both α and β rays, the activity being greatest for rhodium and least for platinum. A large proportion of the β ray activity disappeared in the course of six weeks, and of the α ray activity in one year. It is probable that the two products radium E and F were in part removed with the metals from the radio-lead. We have seen that radium E gives out β rays and loses half of its activity in about six days, while radium F gives out only α rays and its activity falls to half value in 143 days. This conclusion is further confirmed by experiments on the effect of heat on the activity of these substances. By heating to a full red heat, the α ray activity was lost in a few seconds. This is 414in agreement with the results (section 232) where we have seen that radium F is volatilized at about 1000° C. and radium E is left behind.
Salts of gold, silver and mercury added to the radio-lead were found to show only α ray activity on removal. This is in accordance with the view that radium F alone is removed with these substances. Bismuth salts on the other hand showed initially α and β ray activity, but the latter rapidly died away. The presence of β rays in freshly prepared polonium was early observed by Mme Curie. The α and β ray activity of the radio-lead is much reduced by the precipitation of bismuth added to the solution. The α and β ray activity of the radio-lead, however, recovers itself again. This result is exactly what is to be expected if radio-lead contains radium D, E and F. Radium E and F are removed with the bismuth, but the parent substance, radium D, is left behind, and, in consequence, a fresh supply of radium E and F is produced.
While further experiments are required to settle definitely whether the products separated from radio-lead are identical with radium E and F, there can be little doubt that such is the case. This conclusion is strengthened by some experiments which I have made on a specimen of radio-lead, which was kindly forwarded to me by Mr Boltwood of New Haven. This active lead gave out α and β rays, the latter being in unusually large proportion. The active lead was four months old when first tested. The β ray activity in the following six months has remained sensibly constant, but the α ray activity has steadily increased. These results are to be expected if the radio-lead contains radium D. Radium E will reach a practical maximum about 40 days after separation of the product radium D with the lead. The α ray activity due to radium F should increase to a maximum in about 2·6 years (see section 236).
Further experiments are required to settle whether the lead immediately after separation from pitchblende contains only radium D, or whether radium E also appears with it. It seems likely, however, that the bismuth, which is initially present in solution at the time of separation of the lead, will retain both radium E and F, and that the presence of these products in radio-lead is due to their production, after separation, by the parent substance, radium D.
415It would be of scientific value to separate radium D from pitchblende and obtain it in the pure state, for, a month after removal, the β ray activity from it would be about 300 times as great as from an equal weight of radium. By placing a bismuth plate in a solution of this substance, radium F (polonium) should be separated, and, provided a sufficient interval is allowed to elapse, a fresh supply of radium F can at any time be obtained.
The rate of transformation of radium D (half transformed in 40 years) is sufficiently slow not to interfere seriously with its utility in most experiments.
The results of the comparison of the products of radium with those contained in polonium, radio-tellurium and radio-lead are summarized below.
Radium D = product in new radio-lead, no rays. Half transformed in 40 years.
Radium E gives out β rays, separated with bismuth, iridium and platinum. Half transformed in 6 days.
Radium F = product in polonium and radio-tellurium. Gives out only α rays. Half transformed in 143 days.
242. Temporary activity of inactive matter separated from radio-active substances. We have seen in the last section that the platinum metals and bismuth acquire temporary activity by their admixture with a solution of radio-lead, and that these effects are very satisfactorily explained on the view that some of the products of change of radio-lead are removed with the inactive substances. Very similar effects have been observed by Pegram and von Lerch (section 186), when inactive substances were added to solutions of thorium and of the active deposit of thorium. These results, too, are almost certainly due to the removal of one or more of the products of thorium with the inactive matter. Examples of this character may readily be multiplied, and some of the more interesting and important of these will be briefly discussed later.
There have been two general points of view regarding the character of this activity which is temporarily acquired by inactive matter. Some people have supposed that the inactive molecules of the substance, mixed with the solution, acquire by “radio-active induction” temporary activity, the underlying idea being that the close admixture of an inactive and an active substance has communicated 416the property of radiating to some of the molecules of the former. According to the disintegration theory of radio-activity, on the other hand, the temporary activity of originally inactive matter is not due to any alteration of the inactive substance itself, but to an admixture with it of one or more of the numerous radio-active products. The idea of “radio-active induction” has no definite experimental evidence in support of it, while there is much indirect evidence against it.
We shall now consider how these facts are interpreted according to the disintegration theory. In a specimen of old radium, for example, there are present, besides radium itself, the seven successive products which arise from it. Each of these differs in chemical and physical properties from the others. If now, for example, a bismuth rod is introduced into the solution, one or more of these products are deposited on the bismuth. This action is most probably electrolytic in nature, and will depend upon the electro-chemical behaviour of the bismuth compared with that of the products in solution. An electro-negative substance will tend to remove the product or products which are strongly electro-positive. This point of view serves to explain why different metals are made active to different degrees, depending upon their position in the electro-chemical series.
It seems probable that the activity communicated to inactive matter by precipitation from an active solution occurs only during the precipitation. The correctness of this view could readily be tested by observing whether the time that the inactive substance is present in solution has any effect on the magnitude of the activity imparted to it.
When it is remembered that in pitchblende there are present the radio-elements uranium, thorium, radium and actinium and their numerous family of products, it is not surprising that many of the inactive substances separated from it may show very considerable activity due to the mixture of products which may be removed with them. In carrying out experiments on the separation of radium from pitchblende, M. and Mme Curie observed that the separation of the active substance is fairly complete if the stage of purification is not far advanced. Copper, antimony and arsenic can be separated only slightly active, but other substances like lead and iron always show activity. When the stage of 417precipitation is more advanced, every substance separated from the active solution shows activity.
One of the earliest observations in this direction was made by Debierne, who found that barium could be made active by solution with actinium. The active barium removed from the actinium still preserved its activity after chemical treatment, and, in this way, barium chloride was obtained whose activity was 6000 times that of uranium. Although the activity of the barium chloride could be concentrated in the same way as the activity of radiferous barium chloride, it did not show any of the spectroscopic lines of radium, and could not have been due to the admixture of that element with the barium. The activity of the barium was not permanent, and Debierne states that the activity fell to about one-third of its value in three months. It seems probable that the precipitated barium carried down with it the product actinium X, and also some of the actinium itself, and that the decay observed was due to the transformation of actinium X. It is interesting to note that barium is capable of removing a large number of products of the different radio-elements. This effect is probably connected with its position in the electro-chemical series, for barium is highly electro-positive.
Giesel showed in 1900 that bismuth could be made active by placing it in a radium solution, and considered that polonium was in reality bismuth made active by the process of induction. In later experiments, he found that the bismuth plate gave out only α rays, and that the activity of the bismuth could not be ascribed to radium, since no β rays were present. We have seen that this activity of the bismuth is due to the product radium F deposited on its surface.
Mme Curie also found that bismuth was made active by solution with a radium compound, and succeeded in fractionating the above bismuth in the same way as polonium. In this way bismuth was obtained 2000 times as active as uranium, but the activity, like that of polonium separated from pitchblende, decreased with the time. In the light of the experiments on the transformation products of radium, it is seen that these early experiments of Mme Curie add additional confirmation to the view that the product (radium F) separated from radium itself is identical with the polonium obtained directly from pitchblende.
243. It was early recognised that a considerable amount of energy is emitted by the radio-active bodies in the form of their characteristic radiations. Most of the early estimates of the amount of this energy were based on the number and energy of the expelled particles, and were much too small. It has been pointed out (section 114) that the greater part of the energy emitted from the radio-active bodies in the form of ionizing radiations is due to the α rays, and that the β rays in comparison supply only a very small fraction.
Rutherford and McClung[325] made an estimate of the energy of the rays, emitted by a thin layer of active matter, by determining the total number of ions produced by the complete absorption of the α rays. The energy required to produce an ion was determined experimentally by observations of the heating effect of X rays, and of the total number of ions produced when the rays were completely absorbed in air. The energy required to produce an ion in air was found to be 1·90 × 10-10 ergs. This, as will be shown in Appendix A, is probably an over-estimate, but was of the right order of magnitude. From this it was calculated that one gram of uranium oxide spread over a plate in the form of a thin powdered layer emitted energy into the air at the rate of 0·032 gram calories per year. This is a very small emission of energy, but in the case of an intensely radio-active substance like radium, whose activity is about two million times that of uranium, the corresponding emission of energy is 69000 gram calories per year. This is obviously an under-estimate, 419for it includes only the energy radiated into the air. The actual amount of energy released in the form of α rays is evidently much greater than this on account of the absorption of the α rays by the active matter itself.
It will be shown later that the heating effect of radium and of its products is a measure of the energy of the expelled α particles.
244. Heat emission of radium. P. Curie and Laborde[326] first drew attention to the striking result that a radium compound kept itself continuously at a temperature several degrees higher than that of the surrounding atmosphere. Thus the energy emitted from radium can be demonstrated by its direct heating effect, as well as by photographic and electric means. Curie and Laborde determined the rate of the emission of heat in two different ways. In one method the difference of temperature was observed by means of an iron-constantine thermo-couple between a tube containing one gram of radiferous chloride of barium, of activity about ⅙ of pure radium, and an exactly similar tube containing one gram of pure barium chloride. The difference of temperature observed was 1·5° C. In order to measure the rate of emission of heat, a coil of wire of known resistance was placed in the pure barium chloride, and the strength of the electric current required to raise the barium to the same temperature as the radiferous barium was observed. In the other method, the active barium, enclosed in a glass tube, was placed inside a Bunsen calorimeter. Before the radium was introduced, it was observed that the level of the mercury in the stem remained steady. As soon as the radium, which had previously been cooled in melting ice, was placed in the calorimeter, the mercury column began to move at a regular rate. If the radium tube was removed, the movement of the mercury ceased. It was found from these experiments that the heat emission from the 1 gram of radiferous barium, containing about ⅙ of its weight of pure radium chloride, was 14 gram-calories per hour. Measurements were also made with 0·08 gram of pure radium chloride. Curie and Laborde deduced from these results that 1 gram of pure radium emits a quantity of heat equal to about 100 gram-calories per hour. This result was confirmed by the experiments of Runge 420and Precht[327] and others. As far as observation has gone at present, this rate of emission of heat is continuous and unchanged with lapse of time. Therefore, 1 gram of radium emits in the course of a day 2400, and in the course of a year 876,000 gram-calories. The amount of heat evolved in the union of hydrogen and oxygen to form 1 gram of water is 3900 gram-calories. It is thus seen that 1 gram of radium emits per day nearly as much energy as is required to dissociate 1 gram of water.
In some later experiments using 0·7 gram of pure radium bromide, P. Curie[328] found that the temperature of the radium indicated by a mercury thermometer was 3° C. above that of the surrounding air. This result was confirmed by Giesel, who obtained a difference of temperature of 5° C. with 1 gram of radium bromide. The actual rise of temperature observed will obviously depend upon the size and nature of the vessel containing the radium.
During their visit to England in 1903 to lecture at the Royal Institution, M. and Mme Curie performed some experiments with Professor Dewar, to test by another method the rate of emission of heat from radium at very low temperatures. This method depended on the measurement of the amount of gas volatilized when a radium preparation was placed inside a tube immersed in a liquefied gas at its boiling point. The arrangement of the calorimeter is shown in Fig. 97.
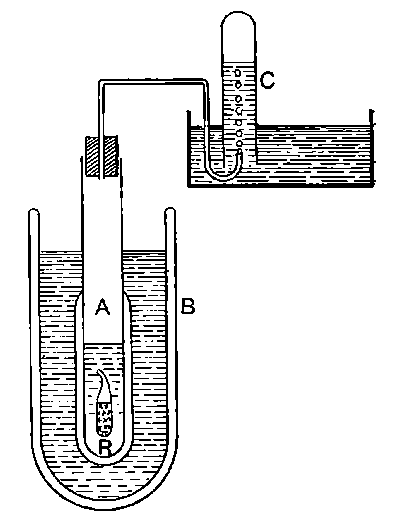
Fig. 97.
The small closed Dewar flask A contains the radium in a glass tube R, immersed in the liquid to be employed. The flask A is surrounded by another Dewar bulb B, containing the same liquid, so that no heat is communicated to A from the outside. The gas liberated in the tube A is collected in the usual way over water or mercury, and its volume determined. By this method, the rate of heat emission of the radium was found to be about the same in 421boiling carbon dioxide and oxygen, and also in liquid hydrogen. Especial interest attaches to the result obtained with liquid hydrogen, for at such a low temperature ordinary chemical activity is suspended. The fact that the heat emission of radium is unaltered over such a wide range of temperature indirectly shows that the rate of expulsion of α particles from radium is independent of temperature, for it will be shown later that the heating effect observed is due to the bombardment of the radium by the α particles.
The use of liquid hydrogen is very convenient for demonstrating the rate of heat emission from a small amount of radium. From 0·7 gram of radium bromide (which had been prepared only 10 days previously) 73 c.c. of gas were given off per minute.
In later experiments P. Curie (loc. cit.) found that the rate of emission of heat from a given quantity of radium depended upon the time which had elapsed since its preparation. The emission of heat was at first small, but after a month’s interval practically attained a maximum. If a radium compound is dissolved and placed in a sealed tube, the rate of heat emission rises to the same maximum as that of an equal quantity of radium in the solid state.
245. Connection of the heat emission with the radiations. The observation of Curie that the rate of heat emission depended upon the age of the radium preparation pointed to the conclusion that the phenomenon of heat emission of radium was connected with the radio-activity of that element. It had long been known that radium compounds increased in activity for about a month after their preparation, when they reached a steady state. It has been shown (section 215), that this increase of activity is due to the continuous production by the radium of the radio-active emanation, which is occluded in the radium compound and adds its radiation to that of the radium proper. It thus seemed probable that the heating effect was in some way connected with the presence of the emanation. Some experiments upon this point were made by Rutherford and Barnes[329]. In order to measure the small amounts of heat emitted, a form of differential air calorimeter shown in Fig. 98 was employed. Two equal glass flasks 422of about 500 c.c. were filled with dry air at atmospheric pressure. These flasks were connected through a glass U-tube filled with xylene, which served as a manometer to determine any variation of pressure of the air in the flasks. A small glass tube, closed at the lower end, was introduced into the middle of each of the flasks. When a continuous source of heat was introduced into the glass tube, the air surrounding it was heated and the pressure was increased. The difference of pressure, when a steady state was reached, was observed on the manometer by means of a microscope with a micrometer scale in the eye-piece. On placing the source of heat in the similar tube in the other flask, the difference in pressure was reversed. In order to keep the apparatus at a constant temperature, the two flasks were immersed in a water-bath, which was kept well stirred.
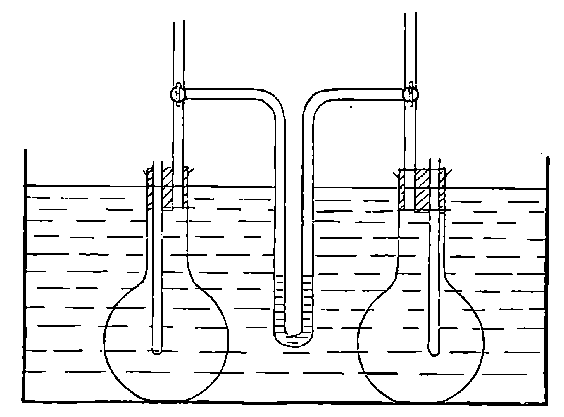
Fig. 98.
Observations were first made on the heat emission from 30 milligrams of radium bromide. The difference in pressure observed on the manometer was standardized by placing a small coil of wire of known resistance in the place of the radium. The strength of the current through the wire was adjusted to give the same difference of pressure on the manometer. In this way it was found that the heat emission per gram of radium bromide corresponded to 65 gram-calories per hour. Taking the atomic weight of radium as 225, this is equivalent to a rate of emission of heat from one gram of metallic radium of 110 gram-calories per hour.
The emanation from the 30 milligrams of radium bromide was then removed by heating the radium (section 215). By passing 423the emanation through a small glass tube immersed in liquid air, the emanation was condensed. The tube was sealed off while the emanation was still condensed in the tube. In this way the emanation was concentrated in a small glass tube about 4 cms. long. The heating effects of the “de-emanated” radium and of the emanation tube were then determined at intervals. It was found that, after removal of the emanation, the heating effect of the radium decayed in the course of a few hours to a minimum, corresponding to about 25 per cent. of the original heat emission, and then gradually increased again, reaching its original value after about a month’s interval. The heating effect of the emanation tube was found to increase for the first few hours after separation to a maximum, and then to decay regularly with the time according to an exponential law, falling to half its maximum value in about four days. The actual heat emission of the emanation tube was determined by sending a current through a coil of wire occupying the same length and position as the emanation tube.
The variation with time of the heating effect from 30 milligrams of radium and the emanation from it is shown in Fig. 99.
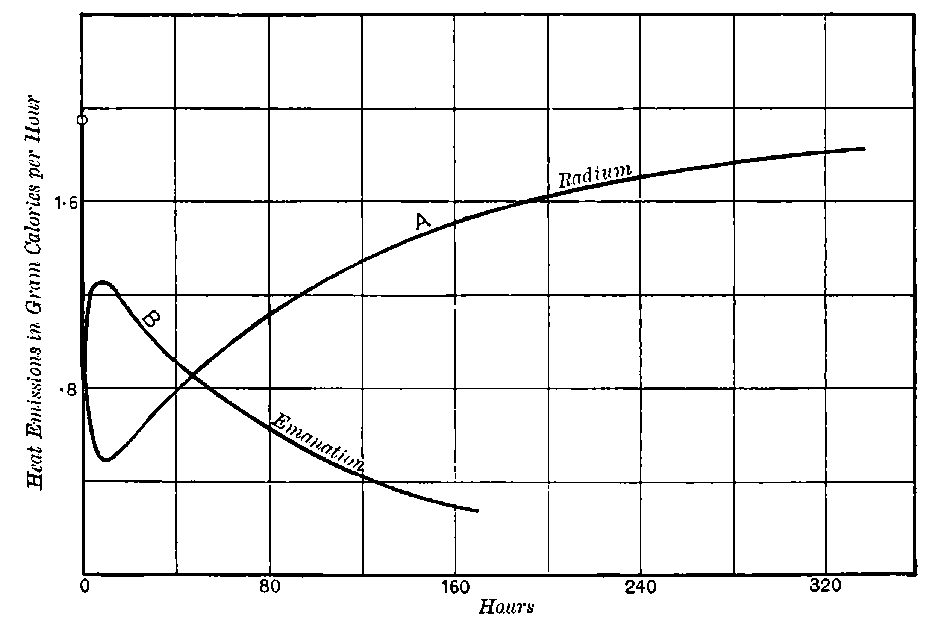
Fig. 99.
Curve A shows the variation with time of the heat emission of the radium and curve B of the emanation. The sum total of 424the rate of heat emission of the radium and the emanation together, was at any time found to be equal to that of the original radium. The maximum heating effect of the tube containing the emanation from 30 milligrams of radium bromide was 1·26 gram-calories per hour. The emanation together with the secondary products which arise from it, obtained from one gram of radium, would thus give out 42 gram-calories per hour. The emanation stored up in the radium is thus responsible for more than two-thirds of the total heat emission from radium. It will be seen later that the decrease to a minimum of the heating effect of radium, after removal of the emanation, is connected with the decay of the excited activity. In a similar way, the increase of the heating effect of the emanation to a maximum some hours after removal is also a result of the excited activity produced by the emanation on the walls of the containing vessel. Disregarding for the moment these rapid initial changes in heat emission, it is seen that the heating effect of the emanation and its further products, after reaching a maximum, decreases at the same rate as that at which the emanation loses its activity, that is, it falls to half value in four days. If Qmax. is the maximum heating effect and Qt the heating effect at any time t later, then
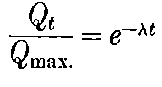
where λ is the constant of change of the emanation.
The curve of recovery of the heating effect of radium from its minimum value is identical with the curve of recovery of its activity measured by the α rays. Since the minimum heating effect is 25 per cent. of the total, the heat emission Qt at any time t after reaching a minimum is given by
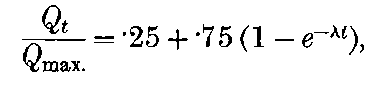
where Qmax. is the maximum rate of heat emission and λ, as before, is the constant of change of the emanation.
The identity of the curves of recovery and fall of the heating effect of radium and its emanation respectively with the corresponding curves for the rise and fall of radio-activity shows that the heat emission of radium and its products is directly connected with their radio-activity. The variation in the heat emission of both radium and its emanation is approximately proportional to their activity measured by the α rays. It is not proportional to 425the activity measured by the β or γ rays, for the intensity of these rays falls nearly to zero some hours after removal of the emanation, while the α ray activity, like the heating effect, is 25 per cent. of the maximum value. These results are thus in accordance with the view that the heat emission of radium accompanies the expulsion of α particles, and is approximately proportional to the number expelled. Before such a conclusion can be considered established, it is necessary to show that the heating effect of the active deposit from the emanation varies in the same way as its α ray activity. Experiments made to test this point will now be considered.
246. Heat emission of the active deposit from the emanation. New radium in radio-active equilibrium contains four successive products which break up with the emission of α particles, viz. radium itself, the emanation, radium A and C. Radium B does not emit rays at all. The effect of the later products radium D, E and F may be neglected, if the radium has not been prepared for more than a year.
It is not easy to settle definitely the relative activity supplied by each of these products when in radio-active equilibrium, but it has been shown in section 229 that the activity is not very different for the four α ray products. The α particles from radium A and C are more penetrating than those from radium itself and the emanation. The evidence at present obtained points to the conclusion that the activity supplied by the emanation is less than that supplied by the other products. This indicates that the α particles from the emanation are projected with less velocity than in the other cases.
When the emanation is suddenly released from radium by heat or solution, the products radium A, B and C are left behind. Since the parent matter is removed, the amount of the products A, B, C at once commences to diminish, and at the end of about three hours reaches a very small value. If the heating effect depends upon the α ray activity, it is thus to be expected that the heat emission of the radium should rapidly diminish to a minimum after the removal of the emanation.
When the emanation is introduced into a vessel, the products radium A, B and C at once appear and increase in quantity, 426reaching a practical maximum about 3 hours later. The heating effect of the emanation tube should thus increase for several hours after the introduction of the emanation.
In order to follow the rapid changes in the heating effect of radium, after removal of the emanation, Rutherford and Barnes (loc. cit.) used a pair of differential platinum thermometers. Each thermometer consisted of 35 cms. of fine platinum wire, wound carefully on the inside of a thin glass tube 5 mms. in diameter, forming a coil 3 cms. long. The glass tube containing the radium and also the tube containing the emanation were selected to slide easily into the interior of the coils, the wire thus being in direct contact with the glass envelope containing the source of heat. The change in resistance of the platinum thermometers, when the radium or emanation tube was transferred from one coil to the other, was readily measured.
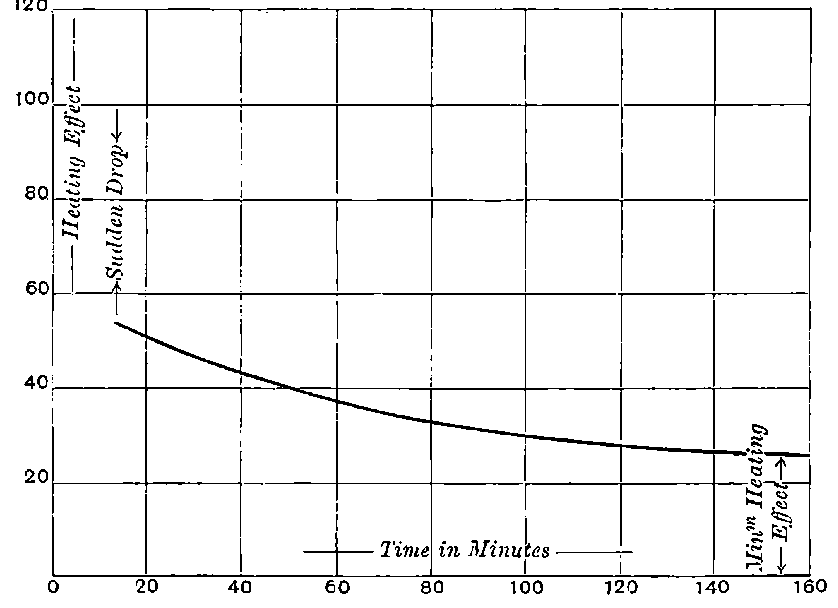
Fig. 100.
The heating effect of the radium in radio-active equilibrium was first accurately determined. The radium tube was heated to drive off the emanation, which was rapidly condensed in a small glass tube 3 cms. long and 3 mms. internal diameter. After allowing a short time for temperature conditions to become steady, the heating effect of the radium tube was measured. The results are shown in Fig. 100. An observation could not be taken until 427about 12 minutes after the removal of the emanation, and the heating effect was then found to have fallen to about 55 per cent. of the maximum value. It steadily diminished with the time, finally reaching a minimum value of 25 per cent. several hours later.
It is not possible in experiments of this character to separate the heating effect of the emanation from that supplied by radium A. Since A is half transformed in three minutes, its heating effect will have largely disappeared after 10 minutes, and the decrease is then mainly due to changes in radium B and C.
The variation with time of the heating effect of the active deposit is still more clearly brought out by an examination of the rise of the heating effect when the emanation is introduced into a small tube, and of the decrease of the heating effect after the emanation is removed. The curve of rise is shown in the upper curve of Fig. 101. 40 minutes after the introduction of the emanation, the heating effect had risen to 75 per cent. of the maximum value which was reached after an interval of about 3 hours.
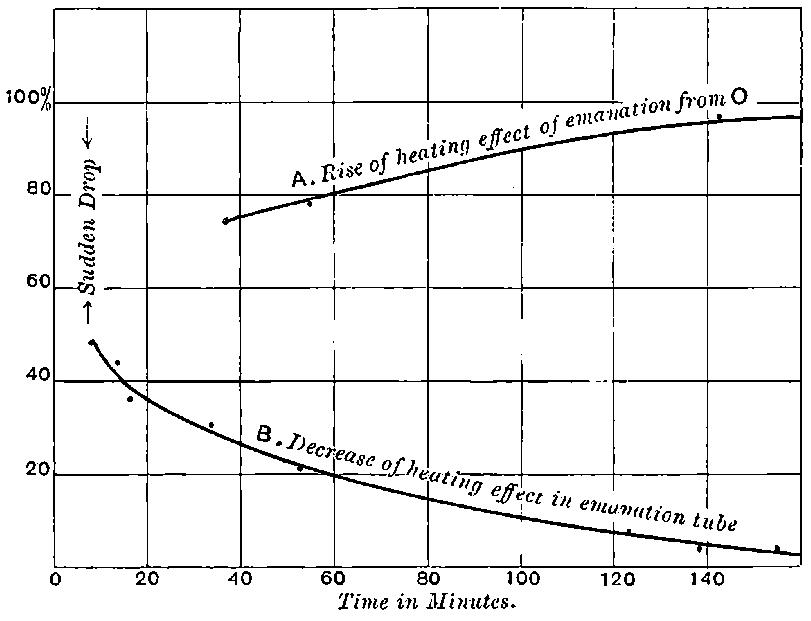
Fig. 101.
After the heating effect of the emanation tube had attained a maximum, the emanation was removed, and the decay with time observed as soon as possible afterwards. The results are shown in the lower curve of Fig. 101. It is seen that the two curves of 428rise and decay are complementary to one another. The first observation was made 10 minutes after removal, and the heating effect had then dropped to 47 per cent. of the original value. This sudden drop is due partly to the removal of the emanation, and partly to the rapid transformation of radium A. The lower curve is almost identical in shape with the corresponding α ray curve for the decay of the excited activity after a long exposure (see Fig. 86) and clearly shows that the heating effect is directly proportional to the activity measured by the α rays over the whole range examined. The heating effect decreases according to the same law and at the same rate as the activity measured by the α rays.
Twenty minutes after the removal of the emanation, radium A has been almost completely transformed, and the activity is then proportional to the amount of radium C present, since the intermediate product B does not give out rays. The close agreement of the activity and heat emission curves shows that the heating effect is proportional also to the amount of radium C. We may thus conclude that the rayless product B supplies little if any of the heat emission observed. If radium B supplied the same amount as radium C, the curve of decrease of heating effect with time would differ considerably from the activity curve.
The conclusion that the transformation of radium B is not accompanied by the release of as much heat as the other changes is to be expected if the heating effect is mainly due to the energy of motion of the expelled α particles.
The relative heating effect due to the radium products is shown in the following table. The initial heating effect of C is deduced by comparison with the corresponding activity curve.
| Products | Radiation | Initial rate of heat emission |
|---|---|---|
| Radium | α rays | 25 per cent. of total |
| Emanation | α „ | |
| Radium A | α „ | 44 „ „ |
| Radium B | no rays | 0 „ „ |
| Radium C | α, β, γ rays | 31 „ „ |
Since radium A and C supply almost an equal proportion of activity, it is probable that they have equal initial heating effects. If this is the case, the heating effect of the emanation alone is 13 per cent. of the total.
429247. Heating effects of the β and γ rays. It has been shown in section 114 that the kinetic energy of the β particles emitted from radium is probably not greater than one per cent. of that due to the α particles. If the heat emission is a result of bombardment by the particles expelled from its mass, it is to be expected that the heating effect of the β rays will be very small compared with that due to the α rays. This anticipation is borne out by experiment. Curie measured the heating effect of radium (1) when enclosed in a thin envelope, and (2) when surrounded by one millimetre of lead. In the former case a large proportion of the β rays escaped, and, in the latter, nearly all were absorbed. The increase of heating effect in case (2) was not more than five per cent., and this is probably an over-estimate.
In a similar way, since the total ionization due to the β rays is about equal to that produced by the γ rays, we should expect that the heating effect of the γ rays will be very small compared with that arising from the α rays.
Paschen made some experiments on the heating effect of radium in a Bunsen ice calorimeter where the radium was surrounded by a thickness of 1·92 cms. of lead—a depth sufficient to absorb a large proportion of the γ rays. In his first publication[330], results were given which indicated that the heating effect of the γ rays was even greater than that of the α rays. This was not confirmed by later observations by the same method. He concluded that the ice calorimeter could not be relied on to measure such very small quantities of heat.
After the publication of Paschen’s first paper Rutherford and Barnes[331] examined the question by a different method. An air calorimeter of the form shown in Fig. 98 was employed which was found to give very satisfactory results. The heat emission of radium was measured (1) when the radium was surrounded by a cylinder of aluminium and (2) when surrounded by a cylinder of lead of the same dimensions. The aluminium absorbed only a small fraction of the γ rays while the lead stopped more than half. No certain difference between the heating effect in the two cases was observed, although from the earlier experiments of Paschen a difference of at least 50 per cent. was to be expected.
430We must therefore conclude that the β and γ rays together do not supply more than a small percentage of the total heat emission of radium—a result which is in accordance with the calculations based on the total ionization produced by the different types of rays.
248. Source of the energy. It has been shown that the heating effect of radium is closely proportional to the activity measured by the α rays. Since the activity is generally measured between parallel plates such a distance apart that most of the α particles are absorbed in the gas, this result shows that the heating effect is proportional to the energy of the emitted α particles. The rapid heat emission of radium follows naturally from the disintegration theory of radio-activity. The heat is supposed to be derived not from external sources, but from the internal energy of the radium atom. The atom is supposed to be a complex system consisting of charged parts in very rapid motion, and in consequence contains a large store of latent energy, which can only be manifested when the atom breaks up. For some reason, the atomic system becomes unstable, and an α particle, of mass about twice that of the hydrogen atom, escapes, carrying with it its energy of motion. Since the α particles would be practically absorbed in a thickness of radium of less than ·001 cm., the greater proportion of the α particles, expelled from a mass of radium, would be stopped in the radium itself and their energy of motion would be manifested in the form of heat. The radium would thus be heated by its own bombardment above the temperature of the surrounding air. The energy of the expelled α particles probably does not account for the whole emission of heat by radium. It is evident that the violent expulsion of a part of the atom must result in intense electrical disturbances in the atom. At the same time, the residual parts of the disintegrated atom rearrange themselves to form a permanently or temporarily stable system. During this process also some energy is probably emitted, which is manifested in the form of heat in the radium itself.
The view that the heat emission of radium is due very largely to the kinetic energy possessed by the expelled α particles is strongly confirmed by calculations of the magnitude of the heating effect to be expected on such an hypothesis. It has been shown 431in section 93 that one gram of radium bromide emits about 1·44 × 1011 α particles per second. The corresponding number for 1 gram of radium (Ra = 225) is 2·5 × 1011. Now it has been calculated from experimental data in section 94, that the average kinetic energy of the α particles expelled from radium is 5·9 × 10-6 ergs. Since all of the α particles are absorbed either in the radium itself or the envelope surrounding it, the total energy of the α particles emitted per second is 1·5 × 106 ergs. This corresponds to an emission of energy of about 130 gram calories per hour. Now the observed heating effect of radium is about 100 gram calories per hour. Considering the nature of the calculation, the agreement between the observed and experimental values is as close as would be expected, and directly supports the view that the heat emission of radium is due very largely to the bombardment of the radium and containing vessel by the α particles expelled from its mass.
249. Heating effect of the radium emanation. The enormous amount of heat liberated in radio-active transformations which are accompanied by the expulsion of α particles is very well illustrated by the case of the radium emanation.
The heat emission of the emanation released from 1 gram of radium is 75 gram calories per hour at its maximum value. This heat emission is not due to the emanation alone, but also to its further products which are included with it. Since the rate of heat emission decays exponentially with the time to about half value in four days, the total amount of heat liberated during the life of the emanation from 1 gram of radium is equal to

since λ = ·0072(hour)-1. Now the volume of the emanation from 1 gram of radium is about 1 cubic millimetre at standard pressure and temperature (section 172). Thus 1 cubic centimetre of the emanation would during its transformation emit 107 gram calories. The heat emitted during the combination of 1 c.c. of hydrogen and oxygen to form water is about 2 gram calories. The emanation thus gives out during its changes 5 × 106 times as much energy as the combination of an equal volume of hydrogen and oxygen 432to form water, although this latter reaction is accompanied by a larger release of energy than any other known to chemistry.
The production of heat from 1 c.c. of the radium emanation is about 21 gram calories per second. This generation of heat would be sufficient to heat to redness, if not to melt down, the walls of the glass tube containing the emanation.
The probable rate of heat emission from 1 gram weight of the emanation can readily be deduced, assuming that the emanation has about 100 times the molecular weight of hydrogen. Since 100 c.c. of the emanation would weigh about 1 gram, the total heat emission from 1 gram of the emanation is about 109 gram calories.
It can readily be calculated that one pound weight of the emanation would, at its maximum, radiate energy at the rate of about 10,000 horse-power. This radiation of energy would fall off with the time, but the total emission of energy during the life of the emanation would correspond to 60,000 horse-power days.
250. Heating effects of uranium, thorium, and actinium. Since the heat emission of radium is a direct consequence of its bombardment by the α particles expelled from its mass, it is to be expected that all the radio-elements which emit α rays should also emit heat at a rate proportional to their α ray activity.
Since the activity of pure radium is probably about two million times that of uranium or thorium, the heat emission from 1 gram of thorium or uranium should be about 5 × 10-5 gram calories per hour, or about 0·44 gram calories per year. This is a very small rate of generation of heat, but it should be detectable if a large quantity of uranium or thorium is employed. Experiments to determine the heating effect of thorium have been made by Pegram[332]. Three kilograms of thorium oxide, enclosed in a Dewar bulb, were kept in an ice-bath, and the difference of temperature between the thorium and ice-bath determined by a set of iron-constantan thermo-electric couples. The maximum difference of temperature observed was 0·04° C., and, from the rate of change of temperature, it was calculated that one gram of thorium oxide liberated 8 × 10-5 gram calories per hour. A more accurate determination of the heat emission is in progress, but the results obtained are of the order of magnitude to be expected.
433251. Energy emitted by a radio-active product. An important consequence follows from the fact that the heat emission is a measure of the energy of the expelled α particles. If each atom of each product emits α particles, the total emission of energy from 1 gram of the product can at once be determined. The α particles from the different products are projected with about the same velocity, and consequently carry off about the same amount of energy. Now it has been shown that the energy of each α particle expelled from radium is about 5·9 × 10-6 ergs. Most of the products probably have an atomic weight in the neighbourhood of 200. Since there are 3·6 × 1019 molecules in one cubic centimetre of hydrogen, it can easily be calculated that there are about 3·6 × 1021 atoms in one gram of the product.
If each atom of the product expels one α particle, the total energy emitted from 1 gram of the matter is about 2 × 1016 ergs or 8 × 108 gram calories. The total emission of energy from a product which emits only β rays is probably about one-hundredth of the above amount.
In this case we have only considered the energy emitted from a single product independently of the successive products which may arise from it. Radium, for example, may be considered a radio-active product which slowly breaks up and gives rise to four subsequent α ray products. The total heat emission from one gram of radium and products is thus about five times the above amount, or 4 × 109 gram calories.
The total emission of energy from radium is discussed later in section 266 from a slightly different point of view.
252. Number of ions produced by an α particle. In the first edition of this book it was calculated by several independent methods that 1 gram of radium emitted about 1011 α particles per second. Since the actual number has later been determined by measuring the charge carried by the α rays (section 93) we can, conversely, use this number to determine with more certainty some of the constants whose values were assumed in the original calculation.
For example, the total number of ions produced by an α 434particle in the gas can readily be determined. The method employed is as follows. 0·484 mgr. of radium bromide was dissolved in water and then spread uniformly over an aluminium plate. After evaporation, the saturation ionization current, due to the radium at its minimum activity, was found to be 8·4 × 10-8 ampere. The plates of the testing vessel were sufficiently far apart to absorb all the α rays in the gas. The number of α particles expelled per second into the gas was found experimentally to be 8·7 × 106. Taking the charge on an ion as 1·13 × 10-19 coulombs (section 36), the total number of ions produced per second in the gas was 7·5 × 1011. Thus each α particle on an average produced 86,000 ions in the gas before it was absorbed.
Now Bragg (section 104) has shown that the α particles from radium at its minimum activity are stopped in about 3 cms. of air. The results obtained by him indicate that the ionization of the particles per cm. of path is less near the radium than some distance away. Assuming, however, as a first approximation that the ionization is uniform along the path, the number of ions produced per cm. of path by the α particle is 29,000. Since the ionization varies directly as the pressure, at a pressure of 1 mm. of mercury the number of ions per unit path would be about 38. Now Townsend (section 103) found that the maximum number of ions produced per unit path of air at 1 mm. pressure by an electron in motion was 20, and in this case a fresh pair of ions was produced at each encounter of the electron with the molecules in its path. In the present case the α particle, which has a very large mass compared with the electron, appears to have a larger sphere of influence than the electron and to ionize twice as many molecules.
In addition, the α particle produces many more ions per unit path than an electron moving with the same velocity, for it has been shown (section 103) that the electron becomes a less efficient ionizer after a certain velocity is reached. As Bragg (loc. cit.) has pointed out, this is to be expected, since the α particle consists of a large number of electrons and consequently would be a far more efficient ionizer than an isolated electron. A calculation of the energy required to produce an ion by an α particle is given in Appendix A.
435 253. Number of β particles expelled from one gram of radium. It is of importance to compare the total number of β particles expelled from one gram of radium in radio-active equilibrium, as, theoretically, this number should bear a definite relation to the total number of α particles emitted. We have seen that new radium in radio-active equilibrium contains four products which emit α rays, viz. radium itself, the emanation, radium A and radium C. On the other hand, β rays are expelled from only one product, radium C. The same number of atoms of each of these successive products in equilibrium break up per second. If the disintegration of each atom is accompanied by the expulsion of one α particle and, in the case of radium C, also of one β particle, the number of α particles emitted from radium in radio-active equilibrium will be four times the number of β particles.
The method employed by Wien to determine the number of β particles emitted from a known quantity of radium has already been discussed in section 80. On account of the absorption of some of the β particles in the radium envelope and in the radium itself, the number found by him is far too small. It has been shown in section 85 that a number of easily absorbed β rays are projected from radium, many of which would be stopped in the radium itself or in the envelope containing it.
In order to eliminate as far as possible the error due to this absorption, in some experiments made by the writer, the active deposit obtained from the radium emanation rather than radium itself was used as a source of β rays. A lead rod, 4 cms. long and 4 mms. in diameter, was exposed as the negative electrode in a large quantity of the radium emanation for three hours. The rod was then removed and the γ ray effect from it immediately measured by an electroscope and compared with the corresponding γ ray effect from a known weight of radium bromide in radio-active equilibrium. Since the active deposit contains the product radium C which alone emits β rays, and, since the intensities of the β and γ rays are always proportional to each other, the number of β particles expelled from the lead rod per second is equal to the corresponding number from the weight of radium bromide which gives the same γ ray effect as the lead rod.
The rod was then enveloped in a thickness of aluminium foil 436of ·0053 cms.—a thickness just sufficient to absorb the α rays—and made the insulated electrode in a cylindrical metal vessel which was rapidly exhausted to a low pressure. The current in the two directions was measured at intervals by an electrometer, and, as we have seen in section 93, the algebraic sum of these currents is proportional to ne, where n is the number of β particles expelled per second from the lead rod, and e the charge on each particle. The activity of the radium C decayed with the time, but, from the known curve of decay, the results could be corrected in terms of the initial value immediately after the rod was removed from the emanation.
Taking into account that half of the β particles emitted by the active deposit were absorbed in the radium itself, and reckoning the charge on the β particle as 1·13 × 10-19 coulombs, two separate experiments gave 7·6 × 1010 and 7·0 × 1010 as the total number of β particles expelled per second from one gram of radium. Taking the mean value, we may conclude that the total number of β particles expelled per second from one gram of radium in radio-active equilibrium is about 7·3 × 1010.
The total number of α particles expelled from one gram of radium at its minimum activity has been shown to be 6·2 × 1010 (section 93). The approximate agreement between these numbers is a strong indication of the correctness of the theoretical views previously discussed. It is to be expected that the number of β particles, deduced in this way, will be somewhat greater than the true value, since the β particles give rise to a secondary radiation consisting also of negatively charged particles moving at a high speed. These secondary β particles, arising from the impact of the β particles on the lead, will pass through the aluminium screen and add their effect to the primary β rays.
The results, however, indicate that four α particles are expelled from radium in radio-active equilibrium for each β particle and thus confirm the theory of successive changes.
254. Theories of radio-activity. In previous chapters, a detailed account has been given of the nature and properties of the radiations, and of the complex processes taking place in the radio-active substances. The numerous products arising from the radio-elements have been closely examined, and have been shown to result from a transformation of the parent element through a number of well-marked stages. In this chapter, the application of the disintegration theory to the explanation of radio-active phenomena will be considered still further, and the logical deductions to be drawn from the theory will be discussed briefly.
A review will first be given of the working hypotheses which have served as a guide to the investigators in the field of radio-activity. These working theories have in many cases been modified or extended with the growth of experimental knowledge.
The early experiments of Mme Curie had indicated that radio-activity was an atomic and not a molecular phenomenon. This was still further substantiated by later work, and the detection and isolation of radium from pitchblende was a brilliant verification of the truth of this hypothesis.
The discovery that the β rays of the radio-elements were similar to the cathode rays produced in a vacuum tube was an important advance, and has formed the basis of several subsequent theories. J. Perrin[333], in 1901, following the views of J. J. Thomson and others, suggested that the atoms of bodies consisted of parts and might be likened to a miniature planetary system. In the 438atoms of the radio-elements, the parts composing the atoms more distant from the centre might be able to escape from the central attraction and thus give rise to the radiation of energy observed. In December 1901, Becquerel[334] put forward the following hypothesis, which, he stated, had served him as a guide in his investigations. According to the view of J. J. Thomson, radio-active matter consists of negatively and positively charged particles. The former have a mass about ¹⁄₁₀₀₀ of the mass of the hydrogen atom, while the latter have a mass about one thousand times greater than that of the negative particle. The negatively charged particles (the β rays) would be projected with great velocity, but the larger positive particles with a much lower velocity forming a sort of gas (the emanation) which deposits itself on the surface of bodies. This in turn would subdivide, giving rise to rays (excited activity).
In a paper communicated to the Royal Society in June 1900, Rutherford and McClung[335] estimated that the energy, radiated in the form of ionizing rays into the gas, was 3000 gram-calories per year for radium of activity 100,000 times that of uranium. Taking the latest estimate of the activity of a pure radium compound as 2,000,000, this would correspond to an emission of energy into the gas in the form of α rays of about 66,000 gram-calories per gram per year. The suggestion was made that this energy might be derived from a re-grouping of the constituents of the atom of the radio-elements, and it was pointed out that the possible energy to be derived from a greater concentration of the components of the atom was large compared with that given out in molecular reactions.
In the original papers[336] giving an account of the discovery of the emanation of thorium and the excited radio-activity produced by it, the view was taken that both of these manifestations were due to radio-active material. The emanation behaved like a gas, while the matter which caused excited activity attached itself to solids and could be dissolved in some acids but not in others. Rutherford and Miss Brooks showed that the radium emanation 439diffused through air like a gas of heavy molecular weight. At a later date Rutherford and Soddy showed that the radium and thorium emanations behaved like chemically inert gases, since they were unaffected by the most drastic physical and chemical treatment.
On the other hand, P. Curie, who, in conjunction with Debierne, had made a series of researches on the radium emanation, expressed dissent from this view. P. Curie[337] did not consider that there was sufficient evidence that the emanation was material in nature, and pointed out that no spectroscopic evidence of its presence had yet been obtained, and also that the emanation disappeared when contained in a sealed vessel. It was pointed out by the writer[338] that the failure to detect spectroscopic lines was probably a consequence of the minute quantity of the emanation present, under ordinary conditions, although the electrical and phosphorescent actions produced by this small quantity are very marked. This contention is borne out by later work. P. Curie at first took the view that the emanation was not material, but consisted of centres of condensation of energy attached to the gas molecules and moving with them.
M. and Mme Curie have throughout taken a very general view of the phenomena of radio-activity, and have not put forward any definite theory. In Jan. 1902, they gave an account of the general working theory[339] which had guided them in their researches. Radio-activity is an atomic property, and the recognition of this fact had created their methods of research. Each atom acts as a constant source of emission of energy. This energy may either be derived from the potential energy of the atom itself, or each atom may act as a mechanism which instantly regains the energy which is lost. They suggested that this energy may be borrowed from the surrounding air in some way not accounted for by the principle of Carnot.
In the course of a detailed study of the radio-activity of thorium, Rutherford and Soddy[340] found that it was necessary to suppose 440that thorium was continuously producing from itself new kinds of active matter, which possess temporary activity and differ in chemical properties from the thorium itself. The constant radio-activity of thorium was shown to be the result of equilibrium between the processes of production of active matter and the change of that already produced. At the same time, the theory was advanced that the production of active matter was a consequence of the disintegration of the atom. The work of the following year was devoted to an examination of the radio-activity of uranium and radium on similar lines, and it was found that the conclusions already advanced for thorium held equally for uranium and radium[341]. The discovery of a condensation of the radio-active emanations[342] gave additional support to the view that the emanations were gaseous in character. In the meantime, the writer[343] had found that the rays consisted of positively charged bodies atomic in size, projected with great velocity. The discovery of the material nature of these rays served to strengthen the theory of atomic disintegration, and at the same time to offer an explanation of the connection between the α rays and the changes occurring in the radio-elements. In a paper entitled “Radio-active Change,” Rutherford and Soddy[344] put forward in some detail the theory of atomic disintegration as an explanation of the phenomena of radio-activity, and at the same time some of the more important consequences which follow from the theory were discussed.
In a paper announcing the discovery of the heat emission of radium, P. Curie and Laborde[345] state that the heat energy may be equally well supposed to be derived from a breaking up of the radium atom or from energy absorbed by the radium from some external source.
J. J. Thomson in an article on “Radium,” communicated to Nature[346], put forward the view that the emission of energy from radium is probably due to some change within the atom, and 441pointed out that a large store of energy would be released by a contraction of the atom.
Sir William Crookes[347], in 1899, proposed the theory that the radio-active elements possess the property of abstracting energy from the gas. If the moving molecules, impinging more swiftly on the substance, were released from the active substance at a much lower velocity, the energy released from the radio-elements might be derived from the atmosphere. This theory was advanced again later on to account for the large heat emission of radium, discovered by P. Curie and Laborde.
F. Re[348] recently advanced a very general theory of matter with a special application to radio-active bodies. He supposes that the parts of the atom were originally free, constituting a nebula of extreme tenuity. These parts have gradually become united round centres of condensation, and have thus formed the atoms of the elements. On this view an atom may be likened to an extinct sun. The radio-active atoms occupy a transitional stage between the original nebula and the more stable chemical atoms, and in the course of their contraction give rise to the heat emission observed.
Lord Kelvin in a paper to the British Association meeting, 1903, has suggested that radium may obtain its energy from external sources. If a piece of white paper is put into one vessel and a piece of black paper into an exactly similar vessel, on exposure of both vessels to the light the vessel containing the black paper is found to be at a higher temperature. He suggests that radium in a similar manner may keep its temperature above the surrounding air by its power of absorption of unknown radiations.
Richarz and Schenck[349] have suggested that radio-activity may be due to the production and breaking up of ozone which is known to be produced by radium salts.
255. Discussion of Theories. From the survey of the general hypotheses advanced as possible explanations of radio-activity, 442it is seen that they may be divided broadly into two classes, one of which assumes that the energy emitted from the radio-elements is obtained at the expense of the internal energy of the atom, and the other that the energy is derived from external sources, but that the radio-elements act as mechanisms capable of transforming this borrowed energy into the special forms manifested in the phenomena of radio-activity. Of these two sets of hypotheses the first appears to be the more probable, and to be best supported by the experimental evidence. Up to the present not the slightest experimental evidence has been adduced to show that the energy of radium is derived from external sources.
J. J. Thomson (loc. cit.) has discussed the question in the following way:—
“It has been suggested that the radium derives its energy from the air surrounding it, that the atoms of radium possess the faculty of abstracting the kinetic energy from the more rapidly moving air molecules while they are able to retain their own energy when in collision with the slowly moving molecules of air. I cannot see, however, that even the possession of this property would explain the behaviour of radium; for imagine a portion of radium placed in a cavity in a block of ice; the ice around the radium gets melted; where does the energy for this come from? By the hypothesis there is no change in the air-radium system in the cavity, for the energy gained by the radium is lost by the air, while heat cannot flow into the cavity from the outside, for the melted ice round the cavity is hotter than the ice surrounding it.”
The writer has recently found that the activity of radium is not altered by surrounding it with a large mass of lead. A cylinder of lead was cast 10 cms. in diameter and 10 cms. high. A hole was bored in one end of the cylinder to the centre, and the radium, enclosed in a small glass tube, was placed in the cavity. The opening was then hermetically closed. The activity was measured by the rate of discharge of an electroscope by the γ rays transmitted through the lead, but no appreciable change was observed during a period of one month.
M. and Mme Curie early made the suggestion that the radiation of energy from the radio-active bodies might be accounted for by supposing that space is traversed by a type of Röntgen rays, and 443that the radio-elements possess the property of absorbing them. Recent experiments (section 279) have shown that there is present at the surface of the earth a very penetrating type of rays, similar to the γ rays of radium. Even if it were supposed that the radio-elements possessed the power of absorbing this radiation, the energy of the rays is far too minute to account even for the energy radiated from an element of small activity like uranium. In addition, all the evidence so far obtained points to the conclusion that the radio-active bodies do not absorb the type of rays they emit to any greater extent than would be expected from their density. It has been shown (section 86) that this is true in the case of uranium. Even if it were supposed that the radio-elements possess the property of absorbing the energy of some unknown type of radiation, which is able to pass through ordinary matter with little absorption, there still remains the fundamental difficulty of accounting for the peculiar radiations from the radio-elements, and the series of changes that occur in them. It is not sufficient for us to account for the heat emission only, for it has been shown (chapter XII) that the emission of heat is directly connected with the radio-activity.
In addition, the distribution of the heat emission of radium amongst the radio-active products which arise from it is extremely difficult to explain on the hypothesis that the energy emitted is borrowed from external sources. It has been shown that more than two-thirds of the heat emitted by radium is due to the emanation together with the active deposit which is produced by the emanation. When the emanation is separated from the radium, its power of emitting heat, after reaching a maximum, decreases with the time according to an exponential law. It would thus be necessary on the absorption hypothesis to postulate that most of the heat emission of radium, observed under ordinary conditions, is not due to the radium itself but to something produced by the radium, whose power of absorbing energy from external sources diminishes with time.
A similar argument also applies to the variation with time of the heating effect of the active deposit produced from the emanation. It has been shown in the last chapter that most of the heating effect observed in radium and its products must be ascribed 444to the bombardment of the α particles expelled from these substances. It has already been pointed out (section 136) that it is difficult to imagine any mechanism, either internal or external, whereby such enormous velocity can suddenly be impressed upon the α particles. We are forced to the conclusion that the α particle did not suddenly acquire this energy of motion, but was initially in rapid motion in the atom, and for some reason, was suddenly released with the velocity which it previously possessed in its orbit.
The strongest evidence against the hypothesis of absorption of external energy is that such a theory ignores the fact, that, whenever radio-activity is observed, it is always accompanied by some change which can be detected by the appearance of new products having chemical properties distinct from those of the original substances. This leads to some form of “chemical” theory, and other results show that the change is atomic and not molecular.
256. Theory of radio-active change. The processes occurring in the radio-elements are of a character quite distinct from any previously observed in chemistry. Although it has been shown that the radio-activity is due to the spontaneous and continuous production of new types of active matter, the laws which control this production are different from the laws of ordinary chemical reactions. It has not been found possible in any way to alter either the rate at which the matter is produced or its rate of change when produced. Temperature, which is such an important factor in altering the rate of chemical reactions, is, in these cases, almost entirely without influence. In addition, no ordinary chemical change is known which is accompanied by the expulsion of charged atoms with great velocity. It has been suggested by Armstrong and Lowry[350] that radio-activity may be an exaggerated form of fluorescence or phosphorescence with a very slow rate of decay. But no form of phosphorescence has yet been shown to be accompanied by radiations of the character of those emitted by the radio-elements. Whatever hypothesis is put forward to explain radio-activity must account not only for the production of a series of active products, which differ in chemical 445and physical properties from each other and from the parent element, but also for the emission of rays of a special character. Besides this, it is necessary to account for the large amount of energy continuously radiated from the radio-elements.
The radio-elements, besides their high atomic weights, do not possess in common any special chemical characteristics which differentiate them from the other elements, which do not possess the property of radio-activity to an appreciable degree. Of all the known elements, uranium, thorium, and radium possess the greatest atomic weights, viz.: radium 225, thorium 232·5, and uranium 240.
If a high atomic weight is taken as evidence of a complicated structure of the atom, it might be expected that disintegration would occur more readily in heavy than in light atoms. At the same time, there is no reason to suppose that the elements of the highest atomic weight must be the most radio-active; in fact, radium is far more active than uranium, although its atomic weight is less. This is seen to be the case also in the radio-active products; for example, the radium emanation is enormously more active weight for weight than the radium itself, and there is every reason to believe that the emanation has an atom lighter than that of radium.
In order to explain the phenomena of radio-activity, Rutherford and Soddy have advanced the theory that the atoms of the radio-elements suffer spontaneous disintegration, and that each disintegrated atom passes through a succession of well-marked changes, accompanied in most cases by the emission of α rays.
A preliminary account of this hypothesis has already been given in section 136, while the mathematical theory of successive changes, which is based upon it, has been discussed in chapter IX. The general theory has been utilized in chapters X and XI to account for the numerous active substances found in uranium, thorium, actinium and radium.
The theory supposes that, on an average, a definite small proportion of the atoms of each radio-active substance becomes unstable at a given time. As a result of this instability, the atoms break up. In most cases, the disintegration is explosive in violence and is accompanied by the ejection of an α particle with 446great velocity; in a few cases, α and β particles are expelled together, while in others a β particle alone escapes. In a few cases, the change in the atom appears to be less violent in character, and is not accompanied by the expulsion of either an α or β particle. The explanation of these rayless changes is considered in section 259. The expulsion of an α particle, of mass about twice that of the hydrogen atom, leaves behind it a new system lighter than the original one, and possessing chemical and physical properties quite different from those of the original element. This new system again becomes unstable, and expels another α particle. The process of disintegration, once started, proceeds from stage to stage at a definite measurable rate in each case.
At any time after the disintegration has commenced, there exists a proportion of the original matter, which is unchanged, mixed with the part which has undergone change. This is in accordance with the observed fact that the spectrum of radium, for example, does not change progressively with time. The radium breaks up so slowly that only a small fraction has been transformed in the course of a few years. The unchanged part still shows its characteristic spectrum, and will continue to do so as long as any radium exists. At the same time it is to be expected that, in old radium, the spectrum of those products which exist in any quantity should also appear.
The term metabolon has been suggested as a convenient expression for each of these changing atoms, derived from the successive disintegration of the atoms of the radio-elements. Each metabolon, on an average, exists only for a limited time. In a collection of metabolons of the same kind the number N, which are unchanged at a time t after production, is given by
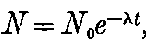
where N₀ is the original number. Now dN/dt = -λN, or the fraction of the metabolons present, which change in unit time, is equal to λ. The value 1/λ may be taken as the average life of each metabolon.
This may be simply shown as follows:—At any time t after N₀ metabolons have been set aside, the number which change in the time dt is equal to λNdt or
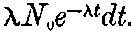
Each 447metabolon has a life t, so that the average life of the whole number is given by
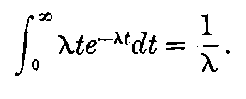
The various metabolons from the radio-elements are distinguished from ordinary matter by their great instability and consequent rapid rate of change. Since a body which is radio-active must ipso facto be undergoing change, it follows that none of the active products, for example, the emanations and Th X, can consist of any known kind of matter; for there is no evidence to show that inactive matter can be made radio-active, or that two forms of the same element can exist, one radio-active and the other not. For example, half of the matter constituting the radium emanation has undergone change after an interval of four days. After the lapse of about one month the emanation as such has nearly disappeared, having been transformed through several stages into other and more stable types of matter, which are in consequence difficult to detect by their radio-activity.
The striking difference in chemical and physical properties which exists in many cases between the various products themselves, and also between the primary active substance and its products, has already been drawn attention to in chapter IX. Some of the products show distinctive electro-chemical behaviour and can be removed from a solution by electrolysis. Others show differences in volatility which have been utilized to effect a partial separation. There can be no doubt that each of these products is a definite new chemical substance, and if it could be collected in sufficient quantity to be examined by ordinary chemical means, would be found to behave like a distinct chemical element. It would differ, however, from the ordinary chemical element in the shortness of its life, and the fact that it is continuously changing into another substance. We shall see later (section 261) that there is every reason to believe that radium itself is a metabolon in the true sense of the term, since it is continuously changing, and is itself produced from another substance. The main point of difference between it and the other products lies in the comparative slowness of its rate of change.
448It is for this reason that radium exists in pitchblende in greater quantity than the other more rapidly changing products. By working up a large amount of the mineral, we have seen that a sufficient quantity of the pure product has been obtained for chemical examination.
On account of the short life of the emanation, it exists in pitchblende in much less quantity than radium, but it, too, has been isolated chemically and its volume measured. The extraordinary properties of this emanation, or gas, have already been discussed, and there can be no doubt that, while it exists, it must be considered a new element allied in chemical properties to the argon-helium group of gases.
There can be no doubt that in the radio-elements we are witnessing the spontaneous transformation of matter, and that the different products which arise mark the stages or halting-places in the process of transformation, where the atoms are able to exist for a short time before again breaking up into new systems.
257. Radio-active products. The following table gives the list of the active products or metabolons known to result from the disintegration of the three radio-elements. In the second column is given the value of the radio-active constant λ for each active product, i.e. the proportion of the active matter undergoing change per second; in the third column the time T required for the activity to fall to one-half, i.e. the time taken for half the active product to undergo change; in the fourth column, the nature of the rays from each active product, not including the rays from the products which result from it; in the fifth column, a few of the more marked physical and chemical properties of each metabolon.
| Products | λ(sec)-1 | T | Nature of the rays | Chemical and Physical properties of the product |
|---|---|---|---|---|
| Uranium | — | — | α | Soluble in excess of ammonium carbonate, soluble in ether. |
| Uranium X | 3·6 × 10-7 | 22 days | β and γ | Insoluble in excess of ammonium carbonate, soluble in ether and water. |
| Thorium | — | — | α | Insoluble in ammonia. |
| Thorium X | 2·0 × 10-6 | 4 days | α | Soluble in ammonia and water. |
| Emanation | 1·3 × 10-2 | 53 secs. | α | Chemically inert gas of heavy molecular weight. Condenses at -120° C. |
| Thorium A | 1·74 × 10-5 | 11 hours | no rays | Deposited on bodies; concentrated on the cathode in an electric field. Soluble in some acids; Th A more volatile than Th B; shows definite electro-chemical behaviour. |
| Thorium B | 2·2 × 10-4 | 55 mins. | α, β, γ | Same |
| ? | — | — | — | |
| Actinium | — | — | no rays | Insoluble in ammonia. |
| Actinium X | 7·8 × 10-7 | 10·2 days | α (and β?) | Soluble in ammonia. |
| Emanation | ·17 | 3·9 secs. | α | Behaves like a gas. |
| Actinium A | 3·2 × 10-4 | 36 mins. | no rays | Deposited on bodies; concentrated on the cathode in an electric field, soluble in ammonia and strong acids; volatilized at a temperature of 100° C., A and B can be separated by electrolysis. |
| Actinium B | 5·4 × 10-3 | 2·15 mins. | α, β, γ | Same |
| ? | — | — | — | |
| Radium | — | 1300 years | α | Allied chemically to barium. |
| Emanation | 2·1 × 10-6 | 3·8 days | α | Chemically inert gas of heavy molecular weight; condenses at -150° C. |
| Radium A (active deposit of rapid change) | 3·85 × 10-3 | 3 mins. | α | } Deposited on surface of bodies; concentrated on cathode in electric field; soluble in strong acids; B volatized at about 700° C., A and C at about 1000° C. |
| Radium B (same) | 5·38 × 10-4 | 21 mins. | no rays | Same |
| Radium C (same) | 4·13 × 10-4 | 28 mins. | α, β, γ | Same |
| Radium D (active deposit of slow change) | — | about 40 | no rays | Soluble in acids; volatile below 1000° C. |
| Radium E (same) | 1·3 × 10-6 | 6 days | β and γ | Non-volatile at 1000° C. |
| Radium F (same) | 5·6 × 10-8 | 143 days | α | Deposited on bismuth from solution; volatile at about 1000° C., same properties as radio-tellurium and polonium. |
The products and their radiations are indicated graphically in Fig. 102 on page 448.
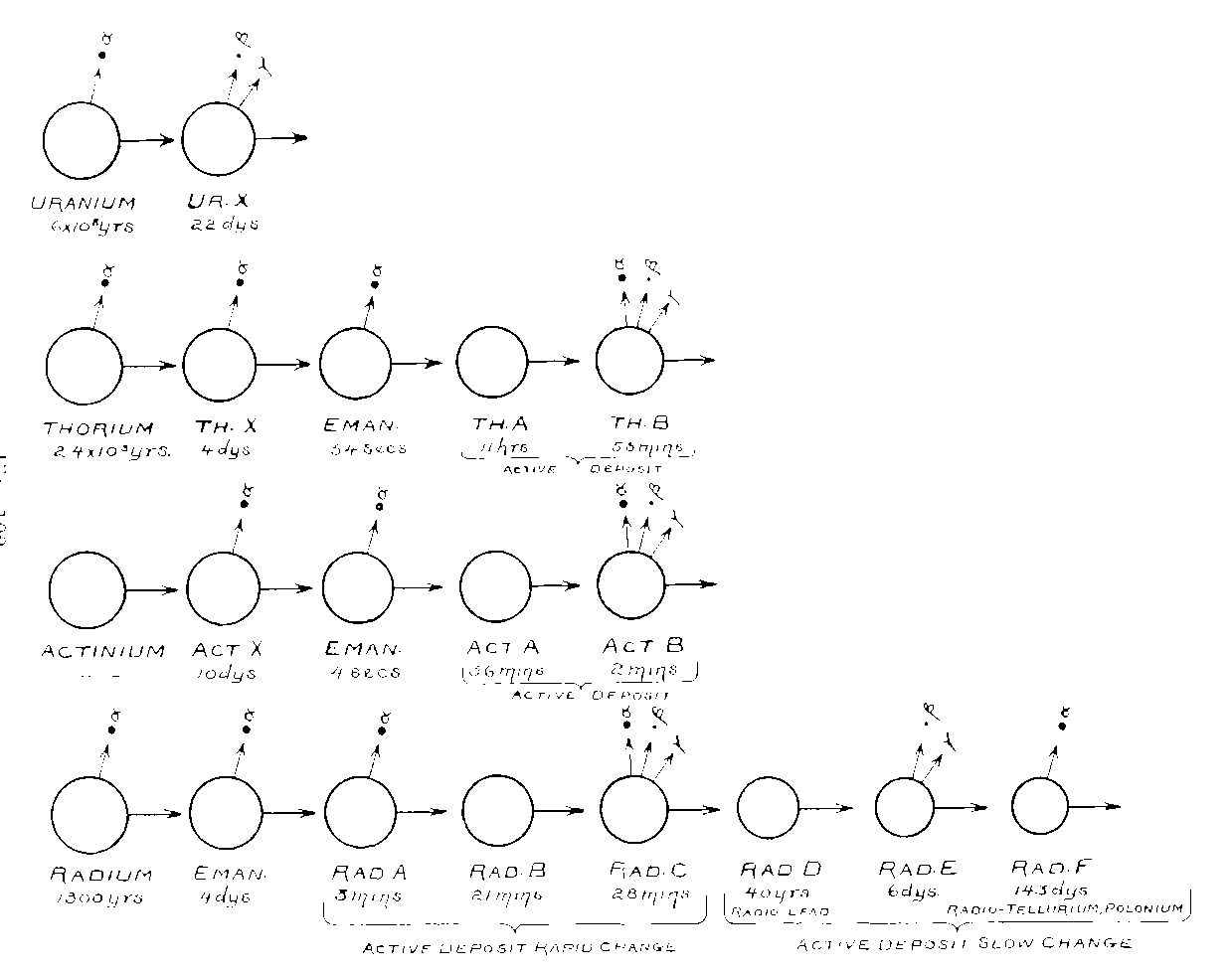
Fig. 102.
One product has been observed in uranium, four in thorium, four in actinium and seven in radium. It is not improbable that a closer examination of the radio-elements may reveal still further changes. If any very rapid transformations exist, they would be very difficult to detect. The change of thorium X into the emanation, for example, would probably not have been discovered if the product of the change had not been gaseous in character. 451The electrolysis of solutions is, in many cases, a very powerful method of separating active products from one another, and its possibilities have not yet been exhausted. The main family of changes of the radio-elements, as far as they are known, have been investigated closely, and it is not likely that any product of comparatively slow rate of change has been overlooked. There is a possibility, however, that two radio-active products may in some cases arise from the disintegration of a single substance. This point is discussed further in section 260.
The remarkable way in which the disintegration theory can be applied to unravel the intricacies of the succession of radio-active changes is very well illustrated in the case of radium. Without its aid, it would not have been possible to disentangle the complicated processes which occur. We have already seen that this analysis has been instrumental in showing that the substances polonium, radio-tellurium and radio-lead are in reality products of radium.
After the radio-active substances have undergone the succession of changes traced above, a final stage is reached where the atoms are either permanently stable, or change so slowly that it is difficult to detect their presence by means of their radio-activity. It is probable, however, that the process of transformation still continues through further slow stages.
There is now considerable evidence that the elements uranium, radium and actinium are intimately connected together. The two latter probably result from the breaking up of uranium. The evidence in support of this idea is given in section 262, but there still remains much work to be done to bridge over the gaps which at present appear to separate these elements from one another.
After the series of transformations have come to an end, there will probably remain a product or products which will be inactive, or active only to a minute extent. In addition, since the α particles, expelled during the transformation, are material in nature, and are non-radio-active, they must collect in some quantity in radio-active matter. The probability that the α particles consist of helium is considered later in section 268.
The value of T, the time for a product to be half-transformed, may be taken as a comparative measure of the stability of the 452different metabolons. The stability of the products varies over a very wide range. For example, the value of T for radium D is 40 years, and for the actinium emanation 3·9 secs. This corresponds to a range of stability measured by 3·8 × 108. The range of stability is still further extended, when it is remembered that the atoms of the radio-elements themselves are very slowly changing.
The only two metabolons of about the same stability are thorium X and the radium emanation. In each case, the transformation is half completed in about four days. I consider that the approximate agreement of the numbers is a mere coincidence, and that the two types of matter are quite distinct from one another; for, if the metabolons were identical, it would be expected that the changes which follow would take place in the same way and at the same rate, but such is not the case. Moreover, Th X and the radium emanation have chemical and physical properties quite distinct from one another.
It is very remarkable that the three radio-active substances, radium, thorium and actinium, should exhibit such a close similarity in the succession of changes which occur in them. Each of them at one stage of its disintegration emits a radio-active gas, and in each case this gas is transformed into a solid which is deposited upon the surface of bodies. It would appear that, after disintegration of an atom of any of these has once begun, there is a similar succession of changes, in which the resulting systems have allied chemical and physical properties. Such a connection is of interest as indicating a possible origin of the recurrence of properties in the atoms of the elements, as exemplified by the periodic law. The connection between thorium and actinium is especially close both as regards the number and nature of the products. The period of transformation of the successive products, though differing in magnitude, rises and falls in a very analogous manner. This indicates that the atoms of these two elements are very similarly constituted.
258. Amount of the products. By application of the theory of successive changes, the probable amount of each of the products present in radium and the other radio-elements can readily be estimated.
453Since each radio-atom expels one α particle of atomic weight about that of hydrogen or helium, the atoms of the intermediate products will not differ much in weight from the parent atom.
The approximate weight of each product present in a gram of radium can be readily deduced. Let NA, NB, NC be the number of atoms of the products A, B, C present per gram in radio-active equilibrium. Let λA, λB, λC be the corresponding constants of change. Then if q is the number of the parent atoms breaking up per second, per gram,
Consider the case of the radium products, where the value of q is 6·2 × 1010 (section 93). Knowing the value of λ and q, the value of N can at once be calculated. The corresponding weight can be deduced, since in one gram of matter of atomic weight about 200, there are about 4 × 1021 atoms (section 39). The results are shown in the following table:—
| Product | Value of λ (sec)-1 | Number of atoms, N, present per gram | Weight of product gram of radium |
|---|---|---|---|
| Radium emanation | 2·0 × 10-6 | 3·2 × 1016 | 8 × 10-3 |
| Radium A | 3·8 × 10-3 | 1·7 × 1013 | 4 × 10-6 |
| Radium B | 5·4 × 10-4 | 1·3 × 1014 | 3 × 10-5 |
| Radium C | 4·1 × 10-4 | 1·6 × 1014 | 4 × 10-5 |
With the small quantities of radium available, the amounts of the products radium A, B and C are too small to weigh. It may be possible, however, to detect their presence by means of the spectroscope.
In the case of thorium, the weight of the product Th X, which is present in greatest quantity, is far too small to be detected. Since the value of λ for Th X is about the same as for the radium emanation, the maximum weight present per gram is about 4 × 10-12 of a gram, remembering that q for radium is about 2 × 106 times the value for thorium. Even with a kilogram of 454thorium, the amount of Th X is far too small to be detected by its weight.
This method can be used generally to calculate the relative amounts of any successive products in radio-active equilibrium, provided the value of λ for each product is known. For example, it will be shown later that uranium is the parent of radium and is half transformed in about 6 × 108 years, while radium and radium D are half transformed in 1300 and 40 years respectively. The weight of radium present in one gram of uranium, when equilibrium is established, is thus 2 × 10-6 grams, and the weight of radium D is 7 × 10-8 grams. In a mineral containing a ton of uranium there should be about 1·8 grams of radium and ·063 grams of radium D. Some recent experiments described in section 262 show that these theoretical estimates are about twice too great.
259. Rayless Changes. The existence of well-marked changes in radium, thorium, and actinium, which are not accompanied by the expulsion of α or β particles, is of great interest and importance.
Since the rayless changes are not accompanied by any appreciable ionization of the gas, their presence cannot be detected by direct means. The rate of change of the substance can, however, be determined indirectly, as we have seen, by measurement of the variation with time of the activity of the succeeding product. The law of change has been found to be the same as for the changes which give rise to α rays. The rayless changes are thus analogous, in some respects, to the monomolecular changes observed in chemistry, with the difference that the changes are in the atom itself, and are not due to the decomposition of a molecule into simpler molecules or into its constituent atoms.
It must be supposed that a rayless change is not of so violent a character as one which gives rise to the expulsion of α or β particles. The change may be accounted for either by supposing that there is a rearrangement of the components of the atom, or that the atom breaks up without the expulsion of its parts with sufficient velocity to produce ionization by collision with the gas. The latter point of view, if correct, at once indicates the possibility that undetected changes of a similar character may be 455taking place slowly in the non-radio-active elements; or, in other words, that all matter may be undergoing a slow process of change. The changes taking place in the radio-elements have been observed only in consequence of the expulsion with great velocity of the parts of the disintegrated atom. Some recent experiments described in Appendix A show that the α particle from radium ceases to ionize the gas when its velocity falls below about 109 cms. per second. It is thus seen that α particles may be projected with a great velocity, and yet fail to produce ionization in the gas. In such a case, it would be difficult to follow the changes by the electrical method, as the electrical effects would be very small in comparison with those produced by the known radio-active bodies.
260. Radiations from the products. We have seen that the great majority of the radio-active products break up with the expulsion of α particles, and that the β particle with its accompaniment of the γ ray appears in most cases only in the last rapid change. In the case of radium, for example, which has been most closely investigated on account of its great activity, radium itself, the emanation and radium A emit only α particles; radium B emits no rays at all; while radium C emits all three kinds of rays. It is difficult to settle with certainty whether the products thorium X and actinium X emit β particles or not, but the β and γ rays certainly appear in each case in the last rapid change in the active deposit, and, in this respect, behave in a similar manner to radium.
The very slow moving electrons which accompany the particles emitted from radium (section 93) are not taken into account, for they appear to be liberated as a result of the impact of α particles on matter, and are expelled with a speed insignificant compared with that of the β particles emitted from radium C.
The appearance of β and γ rays only in the last rapid changes of the radio-elements is very remarkable, and cannot be regarded as a mere coincidence. The final expulsion of a β particle results in the appearance of a product of great stability, or, in the case of radium, of a product (radium D) which has far more stability than the preceding one. It would appear that the initial changes are accompanied by the expulsion of an α particle, and that once 456the β particle is expelled, the components of the residual atom fall into an arrangement of fairly stable equilibrium, where the rate of transformation is very slow. It thus appears probable that the β particle, which is finally expelled, may be regarded as the active agent in promoting the disintegration of the radio-atom through the successive stages. A discussion of this question will be given with more advantage later (section 270), when the general question of the stability of the atom is under consideration.
It is significant that the change in which the three types of rays appear is far more violent in character than the preceding changes. Not only are the α particles expelled with greater velocity than in any other change, but the β particles are projected with a velocity very closely approaching that of light.
There is always a possibility that, in such a violent explosion in the atom, not only may the α and β particles be expelled, but the atom itself may be disrupted into several fragments. If the greater proportion of the matter resulting from the disintegration is of one kind, it would be difficult to detect the presence of a small quantity of rapidly changing matter from observations of the rate of decay; but, if the products have distinctive electro-chemical behaviour, a partial separation should, in some cases, be effected by electrolysis. It has already been pointed out that the results of Pegram and von Lerch (section 207) on the electrolysis of thorium solutions may be explained on the supposition that thorium A and B have distinctive electro-chemical behaviour. Pegram, however, in addition observed the presence of a product which decayed to half value in six minutes. This active product was obtained by electrolysing a solution of pure thorium salt, to which a small quantity of copper nitrate had been added. The copper deposit was slightly active and lost half of its activity in about six minutes.
The presence of such radio-active products, which do not come under the main scheme of changes, indicates that, at some stage of the disintegration, more than one substance results. In the violent disintegration which occurs in radium C and thorium B, such a result is to be expected, for it is not improbable that there are several arrangements whereby the constituents of the atom 457form a system of some slight stability. The two products resulting from the disintegration would probably be present in unequal proportion, and, unless they gave out different kinds of rays, would be difficult to separate from each other.
261. Life of radium. Since the atoms of the radio-elements are continuously breaking up, they must also be considered to be metabolons, the only difference between them and metabolons such as the emanations Th X and others being their comparatively great stability and consequent very slow rate of change. There is no evidence that the process of change, traced above, is reversible under present conditions, and in the course of time a quantity of radium, uranium, or thorium left to itself must gradually be transformed into other types of matter.
There seems to be no escape from this conclusion. Let us consider, for example, the case of radium. The radium is continuously producing from itself the radium emanation, the rate of production being always proportional to the amount of radium present. All the radium must ultimately be changed into emanation, which in turn is transformed through a succession of stages into other kinds of matter. There is no doubt that the emanation is chemically quite different from radium itself. The quantity of radium must diminish, to compensate for the emanation which is formed; otherwise it is necessary to assume that matter in the form of emanation is created from some unknown source.
An approximate estimate of the rate of change of radium can easily be made by two different methods depending upon (1) the number of atoms of radium breaking up per second, and (2) the amount of emanation produced per second.
It has been shown experimentally (section 93) that 1 gram of radium at its minimum activity expels 6·2 × 1010 α particles per second. The heating effect of radium and also its volume agree closely with calculation, if it is supposed that each atom of each product in breaking up emits one α particle. On this supposition it is seen that 6·2 × 1010 atoms of radium break up per second.
Now it has been shown experimentally (section 39) that one cubic centimetre of hydrogen at standard pressure and temperature contains 3·6 × 1019 molecules. Taking the atomic weight of radium 458as 225, the number of atoms in 1 gram of radium is equal to 3·6 × 1021. The fraction λ of radium which breaks up is thus 1·95 × 10-11 per second, or 5·4 × 10-4 per year. It follows that in each gram of radium about half a milligram breaks up per year. The average life of radium is about 1800 years, and half of the radium is transformed in about 1300 years.
We shall now consider the calculation, based on the observed result of Ramsay and Soddy, that the volume of emanation to be obtained from one gram of radium is about 1 cubic millimetre. The experimental evidence based on diffusion results indicates that the molecular weight of the emanation is about 100. If the disintegration theory is correct, the emanation is an atom of radium minus one particle, and therefore must have a molecular weight of at least 200. This high value is more likely to be correct than the experimental number, which is based on evidence that must necessarily be somewhat uncertain. Now the rate of production of emanation per second is equal to λN₀, where N₀ is the equilibrium amount. Taking the molecular weight as 200, the weight of emanation produced per second from 1 gram of radium = 8·96 × 10-6λ = 1·9 × 10-11 gram.
Now the weight of emanation produced per second is very nearly equal to the weight of radium breaking up per second. Thus the fraction of radium breaking up per second is about 1·9 × 10-11, which is in agreement with the number previously calculated by the first method.
We may thus conclude that radium is half transformed in about 1300 years.
Taking the activity of pure radium as about two million times that of uranium, and remembering that only one change, which gives rise to α rays, occurs in uranium and four in radium, it can readily be calculated that the fraction of uranium changing per year is about 10-9. From this it follows that uranium should be half transformed in about 6 × 108 years.
If thorium is a true radio-active element, the time for half transformation is about 2·4 × 109 years, since thorium has about the same activity as uranium but contains four products which emit α rays. If the activity of thorium is due to some radio-active impurity, no estimate of the length of its life can be made until 459the primary active substance has been isolated and its activity measured.
262. Origin of radium. The changes in radium are thus fairly rapid, and a mass of radium if left to itself should in the course of a few thousand years have lost a large proportion of its radio-activity. Taking the above estimate of the life of radium, the value of λ is 5·4 × 10-4, with a year as the unit of time. A mass of radium left to itself should be half transformed in 1300 years and only one-millionth part would remain after 26,000 years. Thus supposing, for illustration, that the earth was originally composed of pure radium, its activity per gram 26,000 years later would not be greater than the activity observed to-day in a good specimen of pitchblende. Even supposing this estimate of the life of radium is too small, the time required for the radium practically to disappear is short compared with the probable age of the earth. We are thus forced to the conclusion that radium is being continuously produced in the earth, unless the very improbable assumption is made, that radium was in some way suddenly formed at a date recent in comparison with the age of the earth. It was early suggested by Rutherford and Soddy[351] that radium might be a disintegration product of one of the radio-elements found in pitchblende. Both uranium and thorium fulfil the conditions required in a possible source of production of radium. Both are present in pitchblende, have atomic weights greater than that of radium, and have rates of change which are slow compared with that of radium. In some respects, uranium fulfils the conditions required better than thorium; for it has not been observed that minerals rich in thorium contain much radium, while on the other hand, the pitchblendes containing the most radium contain a large proportion of uranium.
If radium is not produced from uranium, it is certainly a remarkable coincidence that the greatest activity of pitchblende yet observed is about five or six times that of uranium. Since radium has a life short compared with that of uranium, the amount of radium produced should reach a maximum value after a few thousand years, when the rate of production of fresh radium—which 460is also a measure of the rate of change of uranium—balances the rate of change of that product. In this respect the process would be exactly analogous to the production of the emanation by radium, with the difference that the radium changes much more slowly than the emanation. But since radium itself in its disintegration gives rise to at least five changes with the corresponding production of α rays, the activity due to the radium (measured by the α rays), when in a state of radio-active equilibrium with uranium, should be about five times that of the uranium that produces it; for it has been shown that only one change has so far been observed in uranium in which α rays are expelled. Taking into account the presence of actinium in pitchblende, the activity observed in the best pitchblende is about the same as would be expected if the radium were a disintegration product of uranium. If this hypothesis is correct, the amount of radium in any pitchblende should be proportional to the amount of uranium present, provided the radium is not removed from the mineral by percolating water.
This question has been experimentally attacked by Boltwood[352], McCoy[353] and Strutt[354]. McCoy measured the relative activities of different minerals in the form of powder by means of an electroscope, and determined the amount of uranium present by chemical analysis. His results indicated that the activity observed in the minerals was very approximately proportional to their content of uranium. Since actinium is present as well as uranium and its products, this would indicate that the amount of radium and actinium taken together is proportional to the amount of uranium. This problem has been attacked more directly by Boltwood and Strutt by measuring the relative amount of the radium emanation evolved by different minerals. By dissolving the mineral and then setting it aside in a closed vessel, the amount of emanation present reaches a maximum value after about a month’s interval. The emanation is then introduced into a closed vessel containing a gold-leaf electroscope similar to that shown in Fig. 12. The rate of movement of the gold-leaf is proportional to the amount 461of emanation from the solution, and this in turn is proportional to the amount of radium. Boltwood has made in this way a very complete and accurate comparison of the radium content of different varieties of pitchblende and other ores containing radium. It was found that many of the minerals in the solid state allowed a considerable fraction of the emanation to escape into the air. The percentage fraction of the total amount of emanation lost in this way is shown in Column II of the following table. Column I gives the maximum amount of emanation present in 1 gram of the mineral in arbitrary units when none of the emanation escapes; Column III the weight in grams of uranium contained in 1 gram; and Column IV the ratio obtained by dividing the quantity of emanation by the quantity of uranium. The numbers in Column IV should be constant, if the amount of radium is proportional to the amount of uranium.
| Substance | Locality | I | II | III | IV |
|---|---|---|---|---|---|
| Uraninite | North Carolina | 170·0 | 11·3 | 0·7465 | 228 |
| Uraninite | Colorado | 155·1 | 5·2 | 0·6961 | 223 |
| Gummite | North Carolina | 147·0 | 13·7 | 0·6538 | 225 |
| Uraninite | Joachimsthal | 139·6 | 5·6 | 0·6174 | 226 |
| Uranophane | North Carolina | 117·7 | 8·2 | 0·5168 | 228 |
| Uraninite | Saxony | 115·6 | 2·7 | 0·5064 | 228 |
| Uranophane | North Carolina | 113·5 | 22·8 | 0·4984 | 228 |
| Thorogummite | North Carolina | 72·9 | 16·2 | 0·3317 | 220 |
| Carnotite | Colorado | 49·7 | 16·3 | 0·2261 | 220 |
| Uranothorite | Norway | 25·2 | 1·3 | 0·1138 | 221 |
| Samarskite | North Carolina | 23·4 | 0·7 | 0·1044 | 224 |
| Orangite | Norway | 23·1 | 1·1 | 0·1034 | 223 |
| Euxinite | Norway | 19·9 | 0·5 | 0·0871 | 228 |
| Thorite | Norway | 16·6 | 6·2 | 0·0754 | 220 |
| Fergusonite | Norway | 12·0 | 0·5 | 0·0557 | 215 |
| Aeschynite | Norway | 10·0 | 0·2 | 0·0452 | 221 |
| Xenotine | Norway | 1·54 | 26·0 | 0·0070 | 220 |
| Monazite (sand) | North Carolina | 0·88 | 0·0043 | 205 | |
| Monazite (crys.) | Norway | 0·84 | 1·2 | 0·0041 | 207 |
| Monazite (sand) | Brazil | 0·76 | 0·0031 | 245 | |
| Monazite (massive) | Conn. | 0·63 | 0·0030 | 210 |
With the exception of some of the monazites, the numbers show a surprisingly good agreement, and, taking into consideration the great variation of the content of uranium in the different 462minerals, and the wide range of locality from which they were obtained, the results afford a direct and satisfactory proof that the amount of radium in the minerals is directly proportional to the amount of uranium.
In this connection, it is of interest to note that Boltwood found that a considerable quantity of radium existed in various varieties of monazite, although most of the previous analyses agreed in stating that no uranium was present. A careful examination was in consequence made to test this point, and it was found by special methods that uranium was present, and in about the amount to be expected from the theory. The ordinary methods of analysis failed to give correct results on account of the presence of phosphates. Results of a similar character have recently been given by Strutt[355].
The weight of radium in a mineral per gram of uranium is thus a definite constant of considerable practical importance. Its value was recently determined by Boltwood by comparison of the emanation, liberated from a known weight of uraninite, with that liberated from a known quantity of pure radium bromide, supplied for the purpose by the writer. A measured weight of radium bromide was taken from a stock which gave out heat at a rate of slightly over 100 gram calories per hour per gram, and was thus probably pure. This was dissolved in water, and, by successive dilutions, a standard solution was made up containing 10⁻⁷ gram of radium bromide per c.c. Taking the constitution of radium bromide as RaBr2, it was deduced that the weight of radium per gram of uranium in any mineral was 8·0 × 10⁻⁷ gram. The amount of radium in a mineral per ton of uranium is thus 0·72 gram.
Strutt (loc. cit.) obtained a value nearly twice as great, but he had no means of ascertaining the purity of his radium bromide.
This amount of radium per gram of uranium is of the right order of magnitude to be expected on the disintegration theory, if uranium is the parent of radium. The activity of pure radium, compared with uranium, is not known with sufficient accuracy to 463determine with accuracy the theoretical proportion of radium to uranium.
The production of radium from uranium, while very strongly supported by these experiments, cannot be considered definitely established until direct experimental evidence is obtained of the growth of radium in uranium. The rate of production of radium to be expected on the disintegration theory can readily be estimated. The fraction of uranium breaking up per year has been calculated (section 261) and shown to be about 10-9 per year. This number represents the weight of radium produced per year from 1 gram of uranium. The emanation, released from the amount of radium produced in one year from 1 gram of uranium, would cause an ordinary gold-leaf electroscope to be discharged in about half-an-hour. If a kilogram of uranium is used, the amount of radium produced in a single day should be easily detectable.
Experiments to detect the growth of radium in uranium have been made by several observers. Soddy[356] examined the amount of emanation given off at different times from one kilogram of uranium nitrate in solution, which was originally freed from the small trace of radium present by a suitable chemical process. The solution was kept stored in a closed vessel, and the amount of emanation which collected in the solution was measured at regular intervals.
Preliminary experiments showed that the actual rate of production of radium was far less than the amount to be expected theoretically, and at first very little indication was obtained that radium was produced at all. After allowing the uranium to stand for eighteen months, Soddy states that the amount of emanation was distinctly greater than at first. The solution after this interval contained about 1·5 × 10-9 gram of radium. This gives the value of about 2 × 10-12 for the fraction of uranium changing per year, while the theoretical value is about 10-9.
Whetham[357] also found that a quantity of uranium nitrate which had been set aside for a year showed an appreciable increase in the content of radium, and considers that the rate of production is 464faster than that found by Soddy. In his case, the uranium was not originally completely freed from radium.
Observations extending over years will be required before the question can be considered settled, for the accurate estimation of small quantities of radium by the amount of emanation is beset with difficulties. This is especially the case where observations are made over wide intervals of time.
The writer has made an examination to see if radium is produced from actinium or thorium. It was thought possible that actinium might prove to be an intermediate product between uranium and radium. The solutions, freed from radium, have been set aside for a year, but no certain increase in the content of radium has been observed.
There is little doubt that the production of radium by uranium first proceeds at only a small fraction of the rate to be expected from theory. This is not surprising when we consider that probably several changes intervene between the product Ur X and the radium. In the case of radium, for example, it has been shown that a number of slow changes follow the rapid changes ordinarily observed. On account of the feeble activity of uranium, it would not be easy to detect directly the occurrence of such changes. If, for example, one or more rayless products occurred between Ur X and radium, which were removed from the uranium by the same chemical process used to free it from radium, the rate of production of radium would be very small at first, but would be expected to increase with time as more of the intermediary products were stored up in the uranium. The fact that the contents of uranium and radium in radio-active minerals are always proportional to each other, coupled with definite experimental evidence that radium is produced from uranium, affords an almost conclusive proof that uranium is in some way the parent of radium.
The general evidence which has been advanced to show that radium must be continuously produced from some other substance applies also to actinium, which has an activity of the same order of magnitude as that of radium. The presence of actinium with radium in pitchblende would indicate that this substance also is in some way derived from uranium. It is possible that actinium may prove to be produced either from radium or to be the intermediary 465substance between uranium and radium. If it could be shown that the amount of actinium in radio-active minerals is, like radium, proportional to the amount of uranium, this would afford indirect proof of such a connection. It is not so simple to settle this point for actinium as for radium, since actinium gives out a very short-lived emanation, and the methods adopted to determine the content of radium in minerals cannot be applied without considerable modifications to determine the amount of actinium present.
The experimental data, so far obtained, do not throw much light upon the origin of the primary active matter in thorium. Hofmann and others (section 23) have shown that thorium separated from minerals containing uranium is always more active the greater the quantity of uranium present. This would indicate that the active substance in thorium also may be derived from uranium.
While much work still remains to be done, a promising beginning has already been made in determining the origin and relation of the radio-elements. We have seen that the connection between polonium, radio-tellurium, and radio-lead with radium has already been established. Radium itself is now added to the list, and it is probable that actinium will soon follow.
While the experiments undoubtedly show that there is a definite relation between the amount of uranium and radium present in the ordinary radio-active minerals, Danne[358] has recently called attention to a very interesting apparent exception. Considerable quantities of radium were found in certain deposits in the neighbourhood of Issy-l’Evêque in the Saône-Loire district, although no trace of uranium was present. The active matter is found in pyromorphite (phosphate of lead), in clays containing lead, and in pegmatite, but the radium is usually present in greater quantities in the former. The pyromorphite is found in veins of the quartz and felspar rocks. The veins are always wet owing to the presence of a number of springs in the neighbourhood. The content of uranium in the pyromorphite varies considerably, but Danne considers that about a centigram of radium is present per ton. It seems probable that the radium found in this locality 466has been deposited from water flowing through it, possibly in past times. The presence of radium is not surprising, since crystals of autunite have been found about 40 miles distant, and probably there are deposits containing uranium in that region. This result is of interest, as suggesting that radium may be removed with water and deposited by physical or chemical action some distance away.
It will be shown in the next chapter that radium has been found very widely distributed over the surface of the earth, but generally in very small quantities.
263. Does the radio-activity of radium depend upon its concentration? We have seen that the radio-active constant λ of any product is independent of the concentration of the product. This result has been established over a very wide range for some substances, and especially for the radium emanation. No certain difference in the rate of decay of the emanation has been observed, although the amount present in unit volume of the air has been varied a millionfold.
It has been suggested by J. J. Thomson[359] that the rate of disintegration of radium may be influenced by its own radiations. This, at first sight, appears very probable, for a small mass of a pure radium compound is subjected to an intense bombardment by the radiations arising from it, and the radiations are of such a character that they might be expected to produce a breaking up of the atoms of matter which they traverse. If this be the case, the radio-activity of a given quantity of radium should be a function of its concentration, and should be greater in the solid state than when disseminated through a large mass of matter.
The writer has made an experiment to examine this question. Two glass tubes were taken, in one of which was placed a few milligrams of pure radium bromide in a state of radio-active equilibrium, and in the other a solution of barium chloride. The two tubes were connected near the top by a short cross tube, and the open ends sealed off. The activity of the radium in the solid state was tested immediately after its introduction by placing it in a definite position near an electroscope made of thin metal of the type shown in Fig. 12. The increased rate of discharge of the 467electroscope due to the β and γ rays from the radium was observed. When a lead plate 6 mms. in thickness was placed between the radium and the electroscope, the rate of discharge observed was due to the γ rays alone. By slightly tilting the apparatus, the barium solution flowed into the radium tube and dissolved the radium. The tube was well shaken, so as to distribute the radium uniformly throughout the solution. No appreciable change of the activity measured by the γ rays was observed over the period of one month. The activity measured by the β and γ rays was somewhat reduced, but this was not due to a decrease of the radio-activity, but to an increased absorption of the β rays in their passage through the solution. The volume of the solution was at least 1000 times greater than that of the solid radium bromide, and, in consequence, the radium was subjected to the action of a much weaker radiation. I think we may conclude from this experiment that the radiations emitted by radium have little if any influence in causing the disintegration of the radium atoms.
Voller[360] recently published some experiments which appeared to show that the life of radium varied enormously with its concentration. In his experiments, solutions of radium bromide of known strengths were evaporated down in a platinum vessel 1·2 sq. cms. in area, and their activity tested from time to time. The activity of the radium, so deposited, at first showed the normal rise to be expected on account of the production of the emanation, but after reaching a maximum, it rapidly decayed. For a weight of 10-6 mgrs. of radium bromide, the activity for example, practically disappeared in 26 days after reaching its maximum. The time taken for the activity to disappear increased rapidly with the amount of radium present. In another set of experiments, he states that the activity observed on the vessel was not proportional to the amount of radium present. For example, the activity only increased 24 times for a millionfold increase of the radium present, from 10-9 mgrs. to 10-3 mgrs.
These results, however, have not been confirmed by later experiments made by Eve. He found that, over the range examined, the activity was directly proportional to the amount 468of radium present, within the limits of experimental error. The following table illustrates the results obtained. The radium was evaporated down in platinum vessels 4·9 sq. cms. in area.
| Weight of radium in milligrams | Activity in arbitrary units |
|---|---|
| 10-4 | 1000 |
| 10-5 | 106 |
| 10-6 | 11·8 |
| 10-7 | 1·25 |
For an increase of one-thousandfold of the quantity of radium, the activity increased 800 times, while Voller states that the activity, in his experiments, only increased 3 to 4 times.
In the experiments of Eve, the activity was measured by observing the increased rate of discharge of a gold-leaf electroscope when the platinum vessel containing the active deposit was placed inside the electroscope. The activity of 10-8 mgrs. was too small to be measured with accuracy in the electroscope employed, while 10-3 mgrs. gave too rapid a rate of discharge. On the other hand, the method of measurement employed by Voller was unsuitable for the measurement of very weak radio-activity.
Eve also found that a small quantity of radium kept in a closed vessel did not lose its activity with time. A silvered glass vessel contained a gold-leaf system, such as is shown in Fig. 12. A solution containing 10-6 mgrs. of radium bromide was evaporated over the bottom of the vessel of area 76 sq. cms. The activity, after reaching a maximum, has remained constant over the 100 days during which observations have so far been made.
These experiments of Eve, as far as they go, show that the activity of radium is proportional to the amount of radium present, and that radium, kept in a closed vessel, shows no signs of decreasing in activity. On the other hand, I think there is no doubt that a very small quantity of radium deposited on a plate and left in the open air does lose its activity fairly rapidly. This loss of activity has nothing whatever to do with the shortness of life of the radium itself, but is due to the escape of the radium from the plate into the surrounding gas. Suppose, for example, that a solution containing 10-9 mgrs. of radium bromide is evaporated in a vessel of one sq. cm. in area. This amount of radium is far 469too small to form even a layer of molecular thickness. It seems likely that, during the process of evaporation, the radium would tend to collect in small masses and be deposited on the surface of the vessel. These would very readily be removed by slow currents of air and so escape from the plate. The disappearance of such minute amounts of radium is to be expected, and would probably occur with all kinds of matter present in such minute amount. Such an effect has nothing to do with an alteration of the life of radium and must not be confused with it.
The result that the total radiation from a given quantity of radium depends only on the quantity of radium and not on the degree of its concentration is of great importance, for it allows us to determine with accuracy the content of radium in minerals and in soils in which the radium exists in a very diffused state.
264. Constancy of the radiations. The early observations on uranium and thorium had shown that their radio-activity remained constant over the period of several years during which they were examined. The possibility of separating from uranium and thorium the active products Ur X and Th X respectively, the activity of which decayed with the time, seemed at first sight to contradict this. Further observation, however, showed that the total radio-activity of these bodies was not altered by the chemical processes, for it was found that the uranium and thorium from which the active products were removed, spontaneously regained their radio-activity. At any time after removal of the active product, the sum total of the radio-activity of the separated product together with that of the substance from which it has been separated is always equal to that of the original compound before separation. In cases where active products, like Ur X and the radium emanation, decay with time according to an exponential law, this follows at once from the experimental results. If I1 is the activity of the product at any time t after separation, and I₀ the initial value, we know that
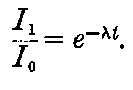
At the same time the activity I2 recovered during the same interval t is given by
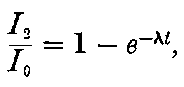
where λ is the same 470constant as before. It thus follows that I1 + I2 = I₀, which is an expression of the above result. The same is also true whatever the law of decay of activity of the separated product (see section 200). For example, the activity of Th X after separation from thorium at first increases with the time. At the same time, the activity of the residual thorium compound at first decreases, and at such a rate that the sum of the activities of the thorium and its separated product is always equal to that of the original thorium.
This apparent constancy of the total radiation follows from the general result that the radio-active processes cannot in any way be changed by the action of known forces. It may be recalled that the constant of decay of the activity of a radio-active product has a definite fixed value under all conditions. It is independent of the concentration of the active matter, of the pressure, and of the nature of the gas in which the substance is placed, and it is not affected by wide ranges of temperature. The only observed exception is the product radium C. Its value of λ increases with temperature to some extent at about 1000° C., but at 1200° C. returns nearly to the normal value. In the same way, it has not been found possible to alter the rate of production of active matter from the radio-elements. In addition, there is not a single well-authenticated case where radio-activity has been altered or destroyed in any active body or created in an inactive element.
Certain cases have been observed, which at first sight seem to indicate a destruction of radio-activity. For example, the excited radio-activity is removed from a platinum wire when heated above a red heat. It has been shown, however, by Miss Gates (section 187) that the radio-activity is not destroyed, but is deposited in unaltered amount on the colder bodies surrounding it. Thorium oxide has been shown to lose to a large extent its power of emanating by ignition to a white heat. But a close examination shows that the emanation is still being produced at the same rate, but is occluded in the compound.
The total radio-activity of a given mass of a radio-element, measured by the peculiar radiations emitted, is a quantity which can neither be increased nor diminished, although it may be manifested in a series of products which are capable of separation from 471the radio-element. The term “conservation of radio-activity” is thus a convenient expression of the facts known at the present time. It is quite possible, however, that further experiments at very high or very low temperatures may show that the radio-activity does vary.
Although no difference has been observed in the radio-activity of uranium over an interval of five years, it has been shown (section 261) that on theoretical grounds the radio-activity of a given quantity of a radio-element should decrease with the time. The change will, however, be so slow in uranium, that probably millions of years must elapse before a measurable change can take place, while the total radio-activity of a given quantity of matter left to itself should thus decrease, but it ought to be constant for a constant mass of the radio-element. It has already been pointed out (section 238) that the activity of radium, measured by the α and β rays, will probably increase for several hundred years after its separation. This is due to the appearance of fresh products in the radium. Ultimately, however, the activity must decrease according to an exponential law with the time, falling to half value in about 1300 years.
The conservation of radio-activity applies not only to the radiations taken as a whole, but also to each specific type of radiation. If the emanation is removed from a radium compound, the amount of β radiation of the radium at once commences to decrease, but this is compensated by the appearance of β rays in the radiations from the vessel in which the separated emanation is stored. At any time the sum total of the β radiations from the radium and the emanation vessel is always the same as that from the radium compound before the emanation was removed.
Similar results have also been found to hold for the γ rays. This was tested by the writer in the following way. The emanation from some solid radium bromide was released by heat, and condensed in a small glass tube which was then sealed off. The radium so treated, and the emanation tube, were placed together under an electroscope, with a screen of lead 1 cm. thick interposed in order to let through only the γ rays. The experiments were continued over three weeks, but the sum total of the γ rays from the radium and the emanation tube was, over the 472whole interval, equal to that of the original radium. During this period the amount of γ rays from the radium at first decreased to only a few per cent. of the original value, and then slowly increased again, until at the end of the three weeks it had nearly regained its original value, before the emanation was removed. At the same time the amount of γ rays from the emanation tube rose from zero to a maximum and then slowly decreased again at the same rate as the decay of the activity of the emanation in the tube. This result shows that the amount of γ rays from radium was a constant quantity over the interval of observation, although the amount of γ rays from the radium and emanation tube had passed through a cycle of changes.
There is one interesting possibility in this connection that should be borne in mind. The rays from the active substances carry off energy in a very concentrated form, and this energy is dissipated by the absorption of the rays in matter. The rays might be expected to cause a disintegration of the atoms of inactive matter on which they fall and thus give rise to a kind of radio-activity. This effect has been looked for by several observers. Ramsay and W. T. Cooke[361] state that they have noticed such an action, using about a decigram of radium as a source of radiation. The radium, sealed in a glass vessel, was surrounded by an external glass tube and exposed to the action of the β and γ rays of radium for several weeks. The inside and outside of the glass tube were found to be active, and the active matter was removed by solution in water. The radio-activity observed was very minute, corresponding to only about 1 milligram of uranium. The writer has, at various times, tried experiments of this character but with negative results. The greatest care is necessary in such experiments to ensure that the radio-activity is not due to other causes besides the rays from the radium. This care is especially necessary in laboratories where considerable quantities of the radium emanation have been allowed to escape into the air. The surface of every substance becomes coated with the slow transformation products of radium, viz. radium D, E, and F. The activity communicated in this way to originally inactive matter is often considerable. This infection by the radium emanation 473extends throughout the whole laboratory, on account of the distribution of the emanation by convection and diffusion. For example, Eve[362] found that every substance which he examined in the laboratory of the writer showed much greater activity than the normal. In this case the radium had been in use in the building for about two years.
265. Loss of weight of the radio-elements. Since the radio-elements are continually throwing off α particles atomic in size, an active substance, enclosed in a vessel sufficiently thin to allow the α particles to escape, must gradually lose in weight. This loss of weight will be small under ordinary conditions, since the greater proportion of the α rays produced are absorbed in the mass of the substance. If a very thin layer of a radium compound were spread on a very thin sheet of substance, which did not appreciably absorb the α particles, a loss of weight due to the expulsion of α particles might be detectable. Since e/m = 6 × 103 for the α particle and e = 1·1 × 10-20 electromagnetic units and 2·5 × 1011 α particles are expelled per second per gram of radium, the proportion of the mass expelled is 4·8 × 10-13 per second and 10-5 per year. There is one condition, however, under which the radium should lose in weight fairly rapidly. If a current of air is slowly passed over a radium solution, the emanation produced would be removed as fast as it was formed. Since the atom of the emanation has a mass probably not much smaller than the radium atom, the fraction of the mass removed per year should be nearly equal to the fraction of the radium which changes per year, i.e. one gram of radium should diminish in weight about half a milligram (section 261) per year.
If it is supposed that the β particles have weight, the loss of weight due to their expulsion is very small compared with that due to the emission of α particles. The writer has shown (section 253) that about 7 × 1010 β particles are projected per second from 1 gram of radium. The consequent loss of weight would only be about 10-9 grams per year.
Except under very special experimental conditions, it would thus be difficult to detect the loss of weight of radium due to 474the expulsion of β particles from its mass. There is, however, a possibility that radium might change in weight even though none of the radio-active products were allowed to escape. For example, if the view is taken that gravitation is the result of forces having their origin in the atom, it is possible that, if the atom were disintegrated, the weight of the parts might not be equal to that of the original atom.
A large number of experiments have been made to see if radium preparations, kept in a sealed tube, alter in weight. With the small quantities of radium available to the experimenter, no difference of weight of radium preparations with time has yet been established with certainty. Heydweiller stated that he had observed a loss of weight of radium and Dorn also obtained a slight indication of change in weight. These results have not, however, been confirmed. Forch, later, was unable to observe any appreciable change.
J. J. Thomson[363] has made experiments to see if the ratio of weight to mass for radium is the same as for inactive matter. We have seen in section 48 that a charge in motion possesses an apparent mass which is constant for slow speeds but increases as the speed of light is approached. Now radium emits some electrons at a velocity comparable with the velocity of light, and presumably these electrons were in rapid motion in the atom before their expulsion. It might thus be possible that the ratio for radium would differ from that for ordinary matter. The pendulum method was used, and the radium was enclosed in a small light tube suspended by a silk fibre. Within the limit of experimental error the ratio of weight to mass was found to be the same as for ordinary matter, so that we may conclude that the number of electrons moving with a velocity approaching that of light is small compared with the total number present.
266. Total emission of energy from the radio-element. It has been shown that 1 gram of radium emits energy at the rate of 100 gram-calories per hour or 876,000 gram-calories per year. If 1 gram of radium in radio-active equilibrium be set apart, its radio-activity and consequent heat emission is given at a 475time t by
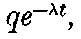
where λ is the constant of decay of activity of radium and of the initial heating effect; the total heat emission from 1 gram of radium is given by
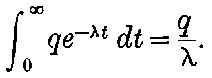
Now on the estimate of the life of radium given in section 261 the value of λ is ¹⁄₁₈₅₀ when 1 year is taken as the unit of time. The total heat emission from 1 gram of radium during its life is thus 1·6 × 109 gram-calories. The heat emitted in the union of hydrogen and oxygen to form 1 gram of water is about 4 × 103 gram-calories, and in this reaction more heat is given out for equal weights than in any other chemical reaction known. It is thus seen that the total energy emitted from 1 gram of radium during its changes is about one million times greater than in any known molecular change. That matter is able, under special conditions, to emit an enormous amount of energy, is well exemplified by the case of the radium emanation. Calculations of the amount of this energy have already been given in section 249.
Since the other radio-elements only differ from radium in the slowness of their change, the total heat emission from uranium and thorium must be of a similar high order of magnitude. There is thus reason to believe that there is an enormous store of latent energy resident in the atoms of the radio-elements. This store of energy could not have been recognized if the atoms had not been undergoing a slow process of disintegration. The energy emitted in radio-active changes is derived from the internal energy of the atoms. The emission of this energy does not disobey the law of the conservation of energy, for it is only necessary to suppose that, when the radio-active changes have ceased, the energy stored up in the atoms of the final products is less than that of the original atoms of the radio-elements. The difference between the energy originally possessed by the matter which has undergone the change, and the final inactive products which arise, is a measure of the total amount of energy released.
There seems to be every reason to suppose that the atomic energy of all the elements is of a similar high order of magnitude. With the exception of their high atomic weights, the radio-elements do not possess any special chemical characteristics which differentiate them from the inactive elements. The existence of 476a latent store of energy in the atoms is a necessary consequence of the modern view developed by J. J. Thomson, Larmor, and Lorentz, of regarding the atom as a complicated structure consisting of charged parts in rapid oscillatory or orbital motion in regard to one another. The energy may be partly kinetic and partly potential, but the mere concentration of the charged particles, which probably constitute the atom, in itself implies a large store of energy in the atom, in comparison with which the energy emitted during the changes of radium is insignificant.
The existence of this store of latent energy does not ordinarily manifest itself, since the atoms cannot be broken up into simpler forms by the physical or chemical agencies at our disposal. Its existence at once explains the failure of chemistry to transform the atoms, and also accounts for the rate of change of the radio-active processes being independent of all external agencies. It has not so far been found possible to alter in any way the rate of emission of energy from the radio-elements. If it should ever be found possible to control at will the rate of disintegration of the radio-elements, an enormous amount of energy could be obtained from a small quantity of matter.
267. Production of helium from radium and the radium emanation. Since the final products, resulting from a disintegration of the radio-elements, are not radio-active, they should in the course of geologic ages collect in some quantity, and should always be found associated with the radio-elements. Now the inactive products resulting from the radio-active changes are the α particles expelled at each stage, and the final inactive product or products which remain, when the process of disintegration can no longer be traced by the property of radio-activity.
Pitchblende, in which the radio-elements are mostly found, contains in small quantity a large proportion of all the known elements. In searching for a possible disintegration product common to all the radio-elements, the presence of helium in the radio-active minerals is noteworthy; for helium is only found in the radio-active minerals, and is an invariable companion of the radio-elements. Moreover, the presence in minerals of a light, inert gas like helium had always been a matter of surprise. The 477production by radium and thorium of the radio-active emanations, which behave like chemically inert gases of the helium-argon family, suggested the possibility that one of the final inactive products of the disintegration of the radio-elements might prove to be a chemically inert gas. The later discovery of the material nature of the α rays added weight to the suggestion; for the measurement of the ratio e/m of the α particle indicated that if the α particle consisted of any known kind of matter, it must either be hydrogen or helium. For these reasons, it was suggested in 1902 by Rutherford and Soddy[364] that helium might be a product of the disintegration of the radio-elements.
Sir William Ramsay and Mr Soddy in 1903 undertook an investigation of the radium emanation, with the purpose of seeing if it were possible to obtain any spectroscopic evidence of the presence of a new substance. First of all, they exposed the emanation to very drastic treatment (section 158), and confirmed and extended the results previously noted by Rutherford and Soddy that the emanation behaved like a chemically inert gas, and in this respect possessed properties analogous to the gases of the helium-argon group.
On obtaining 30 milligrams of pure radium bromide (prepared about three months previously) Ramsay and Soddy[365] examined the gases, liberated by solution of the radium bromide in water, for the presence of helium. A considerable quantity of hydrogen and oxygen was released by the solution (see section 124). The hydrogen and oxygen were removed by passing the liberated gases over a red-hot spiral of partially oxidized copper-wire and the resulting water vapour was absorbed in a phosphorus pentoxide tube.
The gas was then passed into a small vacuum tube which was in connection with a small U tube. By placing the U tube in liquid air, most of the emanation present was condensed, and also most of the CO2 present in the gas. On examining the spectrum of the gas in the vacuum tube, the characteristic line D3 of helium was observed.
478This experiment was repeated with 30 milligrams of radium bromide about four months old, lent for the purpose by the writer. The emanation and CO2 were removed by passing them through a U tube immersed in liquid air. A practically complete spectrum of helium was observed, including the lines of wave-lengths 6677, 5876, 5016, 4972, 4713 and 4472. There were also present three other lines of wave-lengths about 6180, 5695, 5455 which have not yet been identified.
In later experiments, the emanation from 50 milligrams of the radium bromide was conveyed with oxygen into a small U tube, cooled in liquid air, in which the emanation was condensed. Fresh oxygen was added, and the U tube again pumped out. The small vacuum tube, connected with the U tube, showed at first no helium lines when the liquid air was removed. The spectrum obtained was a new one, and Ramsay and Soddy considered it to be probably that of the emanation itself. After allowing the emanation tube to stand for four days, the helium spectrum appeared with all the characteristic lines, and in addition, three new lines present in the helium obtained by solution of the radium. These results have since been confirmed. The experiments, which have led to such striking and important results, were by no means easy of performance, for the quantity of helium and of emanation released from 50 mgrs. of radium bromide is extremely small. It was necessary, in all cases, to remove almost completely the other gases, which were present in sufficient quantity to mask the spectrum of the substance under examination. The success of the experiments has been largely due to the application, to this investigation, of the refined methods of gas analysis, previously employed by Sir William Ramsay with so much skill in the separation of the rare gases xenon and krypton, which exist in minute proportions in the atmosphere. The fact that the helium spectrum was not present at first, but appeared after the emanation had remained in the tube for some days, shows that the helium must have been produced from the emanation. The emanation cannot be helium itself, for, in the first place, helium is not radio-active, and in the second place, the helium spectrum was not present at first, when the quantity of emanation in the tube was at its maximum. Moreover, the diffusion experiments, already discussed, 479point to the conclusion that the emanation is of high molecular weight. There can thus be no doubt that the helium is derived from the emanation of radium in consequence of changes of some kind occurring in it.
These results were confirmed later by other observers. Curie and Dewar[366] performed the following experiment: A weight of about ·42 gr. of radium bromide was placed in a quartz tube, and the tube exhausted until no further gas came off. The radium was then heated to fusion, about 2·6 c.c. of gas being liberated in the process. The tube was then sealed, and some weeks afterwards the spectrum of the gas liberated in the tube by the radium was examined by Deslandres and found to give the entire spectrum of helium. The gas, liberated during the initial heating of the radium, was collected and found to contain a large amount of emanation, although the gas had been passed through two tubes immersed in liquid air. The tube containing these gases was very luminous and rapidly turned violet, while more than half of the gases was absorbed. The spectrum of the phosphorescent light was found to be discontinuous, consisting of three nitrogen bands. No sign of the helium spectrum was observed, although helium must have been present.
Himstedt and Meyer[367] placed 50 mgrs. of radium bromide in a U tube connected with a small vacuum tube. The tube was carefully exhausted and then sealed off. The spectrum of hydrogen and carbon dioxide alone was observed for three months, but after four months the red, yellow, green and blue lines of the helium spectrum were visible. The slow appearance of the helium spectrum was probably due to the presence in the tube of a considerable quantity of hydrogen. In another experiment, some radium sulphate which had been heated to a bright red heat in a quartz tube was connected with a small vacuum tube. After three weeks, some of the lines of helium were clearly seen, and increased in brightness with time.
268. Connection between helium and the α particles. The appearance of helium in a tube containing the radium emanation may indicate either that the helium is one of the final 480products, which appear at the end of the series of radio-active changes, or that the helium is in reality the expelled α particle. The evidence at present points to the latter as being the more probable explanation. In the first place, the emanation diffuses like a gas of heavy molecular weight, and it appears probable that after the expulsion of a few α particles, the atomic weight of the final product is comparable with that of the emanation. On the other hand, the value of e/m determined for the projected α particle points to the conclusion that, if it consists of any known kind of matter, it is either hydrogen or helium.
There has been a tendency to assume that the helium produced from the radium emanation is the last transformation product of that substance. The evidence, however, does not support this view. We have seen that the emanation, after the initial rapid changes, is transformed very slowly. If the helium were the final product, the amount present in the emanation tube after a few days or weeks would be insignificant, since the product radium D intervenes, which takes 40 years to be half transformed. Since the helium cannot be the final product of the series of changes, and since all the other products are radio-active, and almost certainly of high atomic weight, it is difficult to see what position the helium atom occupies in the scheme of transformation, unless it be the α particle expelled during the successive changes.
It is a matter of great difficulty to settle definitely whether the α particle is a projected helium atom or not. On account of the very small deflection of the α rays in an electric field, and the complex nature of the α radiation from radium, an accurate determination of the value e/m for the α particle is beset with difficulties.
It may be possible to settle the question by accurate measurements of the volume of gas in a tube, filled originally with the radium emanation. Since the emanation itself, and two of the rapidly changing products which result from it, emit α particles, the final volume of the α particles, if they can exist in the gaseous state, would be three times the volume of the emanation. Ramsay and Soddy (section 172) have made experiments of this kind, but the results obtained were very contradictory, depending upon the kind of glass employed. In one case, the volume of the 481residual gases shrank almost to zero, in another the initial volume increased to about ten times its initial value. In the latter experiment a brilliant spectrum of helium was observed in the residual gas. This difference of behaviour is probably due to different degrees of absorption of helium by the glass tubes.
If the α particles are helium atoms, we may expect that a large proportion of the helium, which is produced in a tube containing the radium emanation, will be buried in the wall of the glass tube; for the α particles are projected with sufficient velocity to penetrate some distance into the glass. This helium may either remain in the glass, or in some cases rapidly diffuse out again. In any case, a fraction of the helium would be liberated when an intense electric discharge is passed through the tube. Ramsay and Soddy have in some instances observed that a slight amount of helium is liberated on heating the walls of the tube in which the emanation had been stored for some time.
The volume of helium produced per year by 1 gram of radium can easily be calculated on the assumption that the α particle is in reality a helium atom.
It has been shown that 2·5 × 1011 α particles are projected per second from 1 gram of radium. Since there are 3·6 × 1019 molecules in one cubic centimetre of any gas at standard pressure and temperature, the volume of the α particles released per second is 7 × 10-9 c.c. and per year 0·24 c.c. It has already been pointed out that, on this hypothesis, the volume of helium released by the emanation is three times the volume of the latter. The amount of helium to be obtained from the emanation released from 1 gram of radium in radio-active equilibrium is thus about 3 cubic mms.
Ramsay and Soddy have tried to estimate experimentally the probable volume of helium produced per second by one gram of radium. The helium, obtained from 50 mgrs. of radium bromide, which had been kept in solution in a closed vessel for 60 days, was introduced into a vacuum tube. Another similar tube was placed in series with it, and the amount of the helium in the latter adjusted until on passing a discharge through the two tubes in series the helium lines in each tube were of about the same brightness. In this way they calculated that the amount of helium 482present was 0·1 cubic mm. On this estimate, the amount of helium produced per year per gram of radium is about 20 cubic mms. We have seen that the calculated amount is about 240 cubic mms., on the assumption that the α particle is a helium atom. Ramsay and Soddy consider that the presence of argon in one of the tubes may have seriously interfered with the correctness of the estimation. On account of the great uncertainty attaching to estimates of the above character, the value deduced by Ramsay and Soddy does not exclude the probability that the calculated volume may be of the right order of magnitude.
In order to explain the presence of helium in radium on ordinary chemical lines, it has been suggested that radium is not a true element, but a molecular compound of helium with some substance known or unknown. The helium compound gradually breaks down, giving rise to the helium observed. It is at once obvious that this postulated helium compound is of a character entirely different from that of any other compound previously observed in chemistry. Weight for weight, it emits during its change an amount of energy at least one million times greater than any molecular compound known (see section 249). In addition, it must be supposed that the rate of breaking up of the helium compound is independent of great ranges of temperature—a result never before observed in any molecular change. The helium compound in its breaking up must give rise to the peculiar radiations and also pass through the successive radio-active changes observed in radium.
Thus in order to explain the production of helium and radio-activity on this view, a unique kind of molecule must be postulated—a molecule, in fact, which is endowed with every single property which on the disintegration theory is ascribed to the atom of the radio-elements. On the other hand, radium as far as it has been examined, has fulfilled every test required for an element. It has a well-marked and characteristic spectrum, and there is no reason to suppose that it is not an element in the ordinarily accepted sense of the term.
On the theory that the radio-elements are undergoing atomic disintegration, the helium must be considered to be a constituent of the radium atom, or, in other words, the radium atom is 483built up of parts, one of which, at least, is the atom of helium. The theory that the heavy atoms are all built up of some simple fundamental unit of matter or protyle has been advanced at various times by many prominent chemists and physicists. Prout’s hypothesis that all elements are built up out of hydrogen is an example of this point of view of regarding the subject.
On the disintegration theory, the changes occurring in the radio-atoms involve an actual transformation of the atoms through successive changes. This change is so slow in uranium and thorium that at least a million years would be required before the amount of change could be measured by the balance. In radium it is a million times faster, but even in this case it is doubtful whether any appreciable change would have been observed by ordinary chemical methods for many years had not the possibility of such a change been suggested from other lines of evidence.
The similarity of the α particles from the different radio-elements indicates that they consist of expelled particles of the same kind. On this view, helium should be produced by each of the radio-elements. Its presence in minerals containing thorium, for example in monazite sand and the Ceylon mineral described by Ramsay, indicates that helium may be a product of thorium as well as of radium. Strutt[368] has recently suggested that most of the helium observed in radio-active minerals may be a decomposition product of thorium rather than of uranium and radium; for he finds that minerals rich in helium always contain thorium, while many uranium minerals nearly free from thorium contain little helium. The evidence in support of this view is, however, not altogether satisfactory, for some of the uranium minerals in question are secondary uranium minerals (see Appendix B), deposited by the action of water or other agencies at a comparatively late date, and are also, in many cases, highly emanating, and consequently could not be expected to retain more than a fraction of the helium produced in them.
Taking the view that the α particles are projected helium atoms, we must regard the atoms of the radio-elements as compounds of some known or unknown substance with helium. These compounds break up spontaneously, and at a very slow rate even in the 484case of radium. The disintegration takes place in successive stages, and at most of the stages a helium atom is projected with great velocity. This disintegration is accompanied by an enormous emission of energy. The liberation of such a large amount of energy in the radio-active changes at once explains the constancy of the rate of change under the action of any of the physical and chemical agencies at our command. On this view, uranium, thorium and radium are in reality compounds of helium. The helium, however, is held in such strong combination that the compound cannot be broken up by chemical or physical forces, and, in consequence, these bodies behave as chemical elements in the ordinary accepted chemical sense.
It appears not unlikely that many of the so-called chemical elements may prove to be compounds of helium, or, in other words, that the helium atom is one of the secondary units with which the heavier atoms are built up. In this connection it is of interest to note that many of the elements differ in their atomic weight by four—the atomic weight of helium.
If the α particle is a helium atom, at least three α particles must be expelled from uranium (238·5) to reduce its atomic weight to that of radium (225). It is known that five α particles are expelled from radium during its successive transformations. This would make the atomic weight of the final residue 225 – 20 = 205. This is very nearly the atomic weight of lead, 206·5. I have, for some time, considered it probable that lead is the end or final product of radium. The same suggestion has recently been made by Boltwood[369]. This point of view is supported by the fact that lead is always found in small quantity in all uranium minerals, and that the relative proportions of lead and helium in the radio-active minerals are about the same as would be expected if lead and helium were both decomposition products of radium. Dr Boltwood has drawn my attention to the fact that the proportion of lead in many radio-active minerals varies with the content of helium. A mineral rich in helium in nearly all cases contains more lead than a mineral poor in helium. This cannot be considered, at present, more than a speculation, but the facts as they stand are very suggestive.
485269. Age of radio-active minerals. Helium is only found in the radio-active minerals, and this fact, taken in conjunction with the liberation of helium by radium, indicates that the helium must have been produced as a result of the transformation of radium and the other radio-active substances contained in the minerals. Now in a mineral about half the helium is, in many cases, released by heat and the residue by solution. It seems probable that the helium produced throughout the mass of the mineral is mechanically imprisoned in it. Moss[370] found that, by grinding pitchblende in vacuo, helium is evolved, apparently showing that the helium exists in cavities of the mineral. Travers[371] has suggested that, since helium is liberated on heating, the effect may be due to the heat generated by grinding. The escape of the helium from the heated mineral is probably connected with the fact observed by Jaquerod[372] that helium passes through the walls of a quartz tube, heated above 500° C. The substance of the mineral probably possesses a similar property. Travers considers that helium is present in the mineral in a state of supersaturated solid solution, but the facts are equally well explained by assuming that the helium is mechanically imprisoned in the mass of the mineral.
The sudden rise of temperature observed in the mineral fergusonite, at the time the helium is released, has been found to have nothing to do with the presence of helium, for it also takes place in minerals not containing helium. The old view that helium was in a state of chemical combination with the mineral must be abandoned in the light of these more recent experiments.
Since the helium is only released from some minerals by the action of high temperatures and solution, it appears probable that a large proportion of the helium found in the minerals is unable to escape under normal conditions. Thus if the rate of production of helium by the radio-active substance were definitely known, it should be possible to calculate the age of the mineral by observing the volume of helium liberated from it by solution.
In the absence of such definite information, an approximate 486calculation will be made to indicate the order of magnitude of the time that has elapsed since the mineral was formed or was at a temperature low enough to prevent the escape of the helium.
Let us take, for example, the mineral fergusonite, which was found by Ramsay and Travers[373] to evolve 1·81 c.c. of helium. The fergusonite contained about 7 per cent. of uranium. Now uranium in old minerals probably contains about 8 × 10-7 of its weight of radium (see section 262). One gram of the mineral thus contained about 5·6 × 10-8 grams of radium. Now if the α particle is helium, it has been shown that 1 gram of radium produces 0·24 c.c. of helium per year. The volume of helium produced per year in 1 gram of fergusonite is thus 1·3 × 10-8 c.c. Assuming that the rate of production of helium has been uniform, the time required to produce 1·81 c.c. per gram is about 140 million years. If the calculated rate of production of helium by radium is an over-estimate, the time is correspondingly lengthened.
I think that, when the constants required for these calculations are more definitely fixed, this method will probably give fairly trustworthy information as to the probable age of some of the radio-active minerals of the earth’s crust, and indirectly as to the age of the strata in which they are found.
In this connection it is of interest to note that Ramsay[374] found that a Ceylon mineral, thorianite, contained as much as 9·5 c.c. of helium per gram. According to the analysis by Dunstan, this mineral contains about 76 per cent. of thorium and 12 per cent. of uranium. The unusually large amount of helium evolved from this mineral would indicate that it was formed at an earlier date than the fergusonite previously considered.
270. Possible causes of disintegration. In order to explain the phenomena of radio-activity, it has been supposed that a certain small fraction of the radio-atoms undergoes disintegration per second, but no assumptions have been made as to the cause which produces the instability and consequent disintegration. The instability of the atoms may be supposed to be brought about either by the action of external forces or by that of forces inherent 487in the atoms themselves. It is conceivable, for example, that the application of some slight external force might cause instability and consequent disintegration, accompanied by the liberation of a large amount of energy, on the same principle that a detonator is necessary to start some explosives. It has been shown that the number of atoms of any radio-active product which break up per second is always proportional to the number present. This law of change does not throw any light on the question, for it would be expected equally on either hypothesis. It has not been found possible to alter the rate of change of any product by the application of any known physical or chemical forces, unless possibly it is assumed that the force of gravitation which is not under our control may influence in some way the stability of the radio-atoms.
It seems likely therefore that the cause of the disruption of the atoms of the radio-elements and their products resides in the atoms themselves. According to the modern views of the constitution of the atom, it is not so much a matter of surprise that some atoms disintegrate as that the atoms of the elements are so permanent as they appear to be. In accordance with the hypothesis of J. J. Thomson, it may be supposed that the atoms consist of a number of small positively and negatively charged particles in rapid internal movement, and held in equilibrium by their mutual forces. In a complex atom, where the possible variations in the relative motion of the parts are very great, the atom may arrive at such a phase that one part acquires sufficient kinetic energy to escape from the system, or that the constraining forces are momentarily neutralised, so that the part escapes from the system with the velocity possessed by it at the instant of its release.
Sir Oliver Lodge[375] has advanced the view that the instability of the atom may be a result of radiation of energy by the atom. Larmor has shown that an electron, subject to acceleration, radiates energy at a rate proportional to the square of its acceleration. An electron moving uniformly in a straight line does not radiate energy, but an electron, constrained to move in a circular orbit with constant velocity, is a powerful radiator, for in such a case the electron is continuously accelerated towards the centre. Lodge considered 488the simple case of a negatively charged electron revolving round an atom of mass relatively large but having an equal positive charge and held in equilibrium by electrical forces. This system will radiate energy, and, since the radiation of energy is equivalent to motion in a resisting medium, the particle tends to move towards the centre, and its speed consequently increases. The rate of radiation of energy will increase rapidly with the speed of the electron. When the speed of the electron becomes very nearly equal to the velocity of light, according to Lodge, another effect supervenes. It has been shown (section 82) that the apparent mass of an electron increases very rapidly as the speed of light is approached, and is theoretically infinite at the speed of light. There will be at this stage a sudden increase of the mass of the revolving atom, and, on the supposition that this stage can be reached, a consequent disturbance of the balance of forces holding the system together. Lodge considers it probable that, under these conditions, the parts of the system will break asunder and escape from the sphere of one another’s influence.
It seems probable that the primary cause of the disintegration of the atom must be looked for in the loss of energy of the atomic system due to electromagnetic radiation (section 52). Larmor[376] has shown that the condition to be fulfilled in order that a system of rapidly moving electrons may persist without loss of energy is that the vector sum of the accelerations towards the centre should be permanently zero. While a single electron moving in a circular orbit is a powerful radiator of energy, it is remarkable how rapidly the radiation of energy diminishes if several electrons are revolving in a ring. This has recently been shown by J. J. Thomson[377], who examined mathematically the case of a system of negatively electrified corpuscles, situated at equal intervals round the circumference of a circle, and rotating in one plane with uniform velocity round its centre. For example, he found that the radiation from a group of six particles moving with a velocity of ⅒ of the velocity of light is less than one-millionth part of the radiation from a single particle describing the same orbit with the same velocity. When the velocity is ¹⁄₁₀₀ of that of light the amount of radiation 489is only 10-16 that of a single particle moving with the same velocity in the same orbit.
Results of this kind indicate that an atom consisting of a large number of revolving electrons may radiate energy extremely slowly, and yet, finally, this minute but continuous drain of energy from the atom must result either in a rearrangement of its component parts into a new system, or of an expulsion of electrons or groups of electrons from the atom.
Simple models of atoms to imitate the behaviour of polonium in shooting out α particles, and of radium in shooting out β particles have been discussed by Lord Kelvin[378]. It is possible to devise certain stable arrangements of the positively and negatively electrified particles, supposed to constitute an atom, which, on the application of some disturbing force, break up with the expulsion of a part of the system with great velocity.
J. J. Thomson[379] has mathematically investigated the possible stable arrangements of a number of electrons moving about in a sphere of uniform positive electrification. The properties of such a model atom are very striking, and indirectly suggest a possible explanation of the periodic law in chemistry. He has shown that the electrons, if in one plane, arrange themselves in a number of concentric rings; and generally, if they are not constrained to move in one plane, in a number of concentric shells like the coats of an onion.
The mathematical problem is much simplified if the electrons are supposed to rotate in rings in one plane, the electrons in each ring being arranged at equal angular intervals. The ways in which the number of electrons group themselves, for numbers ranging from 60 to 5 at intervals of 5, are shown in the following table:—
| Number of electrons | 60 | 55 | 50 | 45 | 40 | 35 |
|---|---|---|---|---|---|---|
| Number in successive rings | 20 | 19 | 18 | 17 | 16 | 16 |
| 16 | 16 | 15 | 14 | 13 | 12 | |
| 13 | 12 | 11 | 10 | 8 | 6 | |
| 8 | 7 | 5 | 4 | 3 | 1 | |
| 3 | 1 | 1 |
| 490 | ||||||
| Number of electrons | 30 | 25 | 20 | 15 | 10 | 5 |
|---|---|---|---|---|---|---|
| Number in successive rings | 15 | 13 | 12 | 10 | 8 | 5 |
| 10 | 9 | 7 | 5 | 2 | ||
| 5 | 3 | 1 | ||||
In the next table is given the possible series of arrangements of electrons which can have an outer ring of 20:—
| Number of electrons | 59 | 60 | 61 | 62 | 63 | 64 | 65 | 66 | 67 |
|---|---|---|---|---|---|---|---|---|---|
| Number in successive rings | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 | 20 |
| 16 | 16 | 16 | 17 | 17 | 17 | 17 | 17 | 17 | |
| 13 | 13 | 13 | 13 | 13 | 13 | 14 | 14 | 15 | |
| 8 | 8 | 9 | 9 | 10 | 10 | 10 | 10 | 10 | |
| 2 | 3 | 3 | 3 | 3 | 4 | 4 | 5 | 5 |
The smallest number of electrons which can have an outer ring of 20 is 59, while 67 is the greatest.
The various arrangements of electrons can be classified into families, in which the groupings of the electrons have certain features in common. Thus the group of 60 electrons consists of the same arrangement of electrons as the group of 40 with the addition of an outer ring of 20 electrons; the group of 40 is the same as the group of 24 with an additional ring outside; and the group of 24 in turn is the same as the group of 11 with an extra ring. A series of model atoms may be formed in this way, in which each atom is derived from the preceding member by an additional ring of electrons. Such atoms would be expected to possess many properties in common, and would correspond to the elements in the same vertical column of the periodic table of Mendeléef.
Different arrangements of electrons vary widely in stability. Some may acquire an extra electron or two and yet remain stable, others readily lose an electron without disturbing their stability. The former would correspond to an electro-negative atom, the latter to an electro-positive.
Certain arrangements of electrons are stable if the electrons move with an angular velocity greater than a certain value, but 491become unstable when the velocity falls below this value. Four electrons in motion, for example, are stable in one plane, but when the velocity falls below a certain critical value, the system is unstable, and the electrons tend to arrange themselves at the corners of a regular tetrahedron. J. J. Thomson (loc. cit.) applies this property to explain why an atom of radio-active matter breaks up, as follows:—
“Consider now the properties of an atom containing a system of corpuscles (electrons) of this kind. Suppose the corpuscles were originally moving with velocities far exceeding the critical velocity; in consequence of the radiation from the moving corpuscles, their velocity will slowly—very slowly—diminish; when, after a long interval, the velocity reaches the critical velocity, there will be what is equivalent to an explosion of the corpuscles, the corpuscles will move far away from their original position, their potential energy will decrease, while their kinetic energy will increase. The kinetic energy gained in this way might be sufficient to carry the system out of the atom, and we should have, as in the case of radium, a part of the atom shot off. In consequence of the very slow dissipation of energy by radiation the life of the atom would be very long. We have taken the case of the four corpuscles as the type of a system which, like a top, requires for its stability a certain amount of rotation. Any system possessing this property would, in consequence of the gradual dissipation of energy by radiation, give to the atom containing it radio-active properties similar to those conferred by the four corpuscles.”
271. Heat of the sun and earth. It was pointed out by Rutherford and Soddy[380] that the maintenance of the sun’s heat for long intervals of time did not present any fundamental difficulty if a process of disintegration, such as occurs in the radio-elements, were supposed to be taking place in the sun. In a letter to Nature (July 9, 1903) W. E. Wilson showed that the presence of 3·6 grams of radium in each cubic metre of the sun’s mass was sufficient to account for the present rate of emission of energy by the sun. This calculation was based on the estimate of Curie 492and Laborde that 1 gram of radium emits 100 gram-calories per hour, and on the observation of Langley that each square centimetre of the sun’s surface emits 8·28 × 106 gram-calories per hour. Since the average density of the sun is 1·44, the presence of radium in the sun, to the extent of 2·5 parts by weight in a million, would account for its present rate of emission of energy.
An examination of the spectrum of the sun has not so far revealed any of the radium lines. It is known, however, from spectroscopic evidence that helium is present, and this indirectly suggests the existence of radio-active matter also. It can readily be shown[381] that the absence of penetrating rays from the sun at the surface of the earth does not imply that the radio-elements are not present in the sun. Even if the sun were composed of pure radium, it would hardly be expected that the γ rays emitted would be appreciable at the surface of the earth, since the rays would be almost completely absorbed in passing through the atmosphere, which corresponds to a thickness of 76 centimetres of mercury.
In the Appendix E of Thomson and Tait’s Natural Philosophy, Lord Kelvin has calculated the energy lost in the concentration of the sun from a condition of infinite dispersion, and concludes that it seems “on the whole probable that the sun has not illuminated the earth for 100,000,000 years and almost certain that he has not done so for 500,000,000 years. As for the future we may say, with equal certainty, that inhabitants of the earth cannot continue to enjoy the light and heat essential to their life for many million years longer, unless sources now unknown to us are prepared in the great storehouses of creation.”
The discovery that a small mass of a substance like radium can emit spontaneously an enormous quantity of heat renders it possible that this estimate of the age of the sun’s heat may be much increased. In a letter to Nature (Sept. 24, 1903) G. H. Darwin drew attention to this probability, and at the same time pointed out that, on Kelvin’s hypotheses, his estimate of the duration of the sun’s heat was probably much too high, and stated that, “The lost energy of the sun, supposed to be a homogeneous sphere of mass M and radius a, is (⅗)μM2/a where μ is the constant of 493gravitation. On introducing numerical values for the symbols in this formula, I find the lost energy to be 2·7 × 107 M calories where M is expressed in grams. If we adopt Langley’s value of the solar constant, this heat suffices to give a supply for 12 million years. Lord Kelvin used Pouillet’s value for that constant, but if he had been able to use Langley’s, his 100 million would have been reduced to 60 million. The discrepancy between my results of 12 million and his of 60 million is explained by a conjectural augmentation of the lost energy to allow for the concentration of the solar mass towards its central parts.” Now it has been shown (section 266) that one gram of radium emits during its life an amount of heat corresponding to 1·6 × 109 gram-calories. It has also been pointed out that there is every reason to suppose that a similar amount of energy is resident in the chemical atoms of the inactive elements. It is not improbable that, at the enormous temperature of the sun, the breaking up of the elements into simpler forms may be taking place at a more rapid rate than on the earth. If the energy resident in the atoms of the elements is thus available, the time during which the sun may continue to emit heat at the present rate may be at least 50 times longer than the value computed from dynamical data.
Similar considerations apply to the question of the age of the earth. A full discussion of the probable age of the earth, computed from its secular cooling from a molten mass, is given by Lord Kelvin in Appendix D of Thomson and Tait’s Natural Philosophy. He has there shown that about 100 million years after the earth was a molten mass, the gradual cooling due to radiation from its surface would account for the average temperature gradient of ¹⁄₅₀° F. per foot, observed to-day near the earth’s surface.
Some considerations will now be discussed which point to the probability that the present temperature gradient observed in the earth cannot be used as a guide to estimate the length of time that has elapsed since the earth has been at a temperature capable of supporting animal and vegetable life; for it will be shown that probably there is sufficient radio-active matter on the earth to supply as much heat to the earth as is lost by radiation from its surface. Taking the average conductivity K of the materials of 494the earth as ·004 (C.G.S. units) and the temperature gradient T near the surface as ·00037° C. per cm., the heat Q in gram-calories conducted to the surface of the earth per second is given by
where R is the radius of the earth.
Let X be the average amount of heat liberated per second per cubic centimetre of the earth’s volume owing to the presence of radio-active matter. If the heat Q radiated from the earth is equal to the heat supplied by the radio-active matter in the earth,
Substituting the values of these constants,
Since 1 gram of radium emits 876,000 gram-calories per year, the presence of 2·6 × 10-13 grams of radium per unit volume, or 4·6 × 10-14 grams per unit mass, would compensate for the heat lost from the earth by conduction.
Now it will be shown in the following chapter that radio-active matter seems to be distributed fairly uniformly through the earth and atmosphere. In addition, it has been found that all substances are radio-active to a feeble degree, although it is not yet settled whether this radio-activity may not be due mainly to the presence of a radio-element as an impurity. For example, Strutt[382] observed that a platinum plate was about ¹⁄₃₀₀₀ as active as a crystal of uranium nitrate, or about 2 × 10-10 as active as radium. This corresponds to a far greater activity than is necessary to compensate for the loss of heat of the earth. A more accurate deduction, however, can be made from data of the radio-activity exhibited by matter dug out of the earth. Elster and Geitel[383] filled a dish of 495volume 3·3 × 103 c.c. with clay dug up from the garden, and placed it in a vessel of 30 litres capacity in which was placed an electroscope to determine the conductivity of the enclosed gas. After standing for several days, they found that the conductivity of the air reached a constant maximum value, corresponding to three times the normal. It will be shown later (section 284) that the normal conductivity observed in sealed vessels corresponds to the production of about 30 ions per c.c. per second. The number of ions produced per second in the vessel by the radio-active earth was thus about 2 × 106. This would give a saturation current through the gas of 2·2 × 10-14 electromagnetic units. Now the emanation from 1 gram of radium stored in a metal cylinder gives a saturation current of about 3·2 × 10-5 electromagnetic units. Elster and Geitel considered that most of the conductivity observed in the gas was due to a radio-active emanation, which gradually diffused from the clay into the air in the vessel. The increased conductivity in the gas observed by Elster and Geitel would thus be produced by the emanation from 7 × 10-10 gram of radium. Taking the density of clay as 2, this corresponds to about 10-13 gram of radium per gram of clay. But it has been shown that if 4·6 × 10-14 gram of radium were present in each gram of earth, the heat emitted would compensate for the loss of heat of the earth by conduction and radiation. The amount of activity observed in the earth is thus about the right order of magnitude to account for the heat emission required. In the above estimate, the presence of uranium and thorium minerals in the earth has not been considered. Moreover, it is probable that the total amount of radio-activity in the clay was considerably greater than that calculated, for it is likely that other radio-active matter was present which did not give off an emanation.
If the earth is supposed to be in a state of thermal equilibrium in which the heat lost by radiation is supplied from radio-active matter, there must be an amount of radio-active matter in the earth corresponding to about 270 million tons of radium. If there were more radium than this in the earth, the temperature gradient would be greater than that observed to-day. This may appear to be a very large quantity of radium, but recent determinations (section 281) of the amount of radium emanation in the atmosphere 496strongly support the view that a large quantity of radium must exist in the surface soil of the earth. Eve found, on a minimum estimate, that the amount of emanation always present in the atmosphere is equivalent to the equilibrium amount derived from 100 tons of radium. There is every reason to believe that the emanation found in the atmosphere is supplied both by the diffusion of the emanation from the soil and by the action of springs. Since the emanation loses half its activity in four days, it cannot diffuse from any great depth. Assuming that the radium is uniformly distributed throughout the earth, the quantity of the radium emanation produced in a thin shell of earth about thirteen metres in depth, is sufficient to account for the amount ordinarily observed in the atmosphere.
I think we may conclude that the present rate of loss of heat of the earth might have continued unchanged for long periods of time in consequence of the supply of heat from radio-active matter in the earth. It thus seems probable that the earth may have remained for very long intervals of time at a temperature not very different from that observed to-day, and that, in consequence, the time during which the earth has been at a temperature capable of supporting the presence of animal and vegetable life may be very much longer than the estimate made by Lord Kelvin from other data.
272. Evolution of matter. Although the hypothesis that all matter is composed of some elementary unit of matter or protyle has been advanced as a speculation at various times by many prominent physicists and chemists, the first definite experimental evidence showing that the chemical atom was not the smallest unit of matter was obtained in 1897 by J. J. Thomson in his classic research on the nature of the cathode rays produced by an electric discharge in a vacuum tube. We have seen that Sir William Crookes, who was the first to demonstrate the remarkable properties of these rays, had suggested that they consisted of streams of projected charged matter and represented—as he termed it—a new or “fourth state of matter.”
J. J. Thomson showed by two distinct methods (section 50), 497that the cathode rays consisted of a stream of negatively charged particles projected with great velocity. The particles behaved as if their mass was only about ¹⁄₁₀₀₀ of the mass of the atom of hydrogen, which is the lightest atom known. These corpuscles, as they were termed by Thomson, were found at a later date to be produced from a glowing carbon filament and from a zinc plate exposed to the action of ultra-violet light. They acted as isolated units of negative electricity, and, as we have seen, may be identified with the electrons studied mathematically by Larmor and Lorentz. Not only were these electrons produced by the action of light, heat, and the electric discharge, but similar bodies were also found to be emitted spontaneously from the radio-elements with a velocity far greater than that observed for the electrons in a vacuum tube.
The electrons produced in these various ways were all found to carry a negative charge, and to be apparently identical; for the ratio e/m of the charge of the electron to its mass was in all cases the same within the limits of experimental error. Since electrons, produced from different kinds of matter and under different conditions, were in all cases identical, it seemed probable that they were a constituent part of all matter. J. J. Thomson suggested that the atom is built up of a number of these negatively charged electrons combined in some way with corresponding positively charged bodies.
On this view the atoms of the chemical elements differ from one another only in the number and arrangement of the component electrons.
The removal of an electron from the atom in the case of ionization does not appear to affect permanently the stability of the system, for no evidence has so far been obtained to show that the passage of an intense electric discharge through a gas results in a permanent alteration of the structure of the atom. On the other hand, in the case of the radio-active bodies, a positively charged particle of mass about twice that of the hydrogen atom escapes from the heavy radio-atom. This loss appears to result at once in a permanent alteration of the atom, and causes a marked change in its physical and chemical properties. In addition there is no evidence that the process is reversible.
498The expulsion of a β particle with great velocity from an atom of radio-active matter also results in a transformation of the atom. For example radium E emits a β particle, and, in consequence, gives rise to a distinct substance radium F (polonium). A case of this kind, where the expulsion of a β particle with great velocity causes a complete rearrangement of the parts of an atom, is probably quite distinct from the process which occurs during ionization, where a slow speed electron escapes from the atom without apparently affecting the stability of the atom left behind.
The only direct experimental evidence of the transformation of matter has been derived from a study of the radio-active bodies. If the disintegration theory, advanced to account for the phenomena of radio-activity, is correct in the main essentials, then the radio-elements are undergoing a spontaneous and continuous process of transformation into other and different kinds of matter. The rate of transformation is slow in uranium and thorium, but is fairly rapid in radium. It has been shown that the fraction of a mass of radium which is transformed per year is about ¹⁄₂₀₀₀ of the total amount present. In the case of uranium and thorium probably a million years would be required to produce a similar amount of change. Thus the process of transformation in uranium and thorium is far too slow to be detected within a reasonable time by the use of the balance or spectroscope, but the radiations which accompany the transformation can easily be detected. Although the process of change is slow it is continuous, and in the course of ages the uranium and thorium present in the earth must be transformed into other types of matter.
Those who have considered the possibility of atoms undergoing a process of transformation have generally thought that the matter as a whole would undergo a progressive change, with a gradual alteration of physical and chemical properties of the whole mass of substance. On the theory of disintegration this is not the case. Only a minute fraction of the matter present breaks up in unit time, and in each of the successive stages through which the disintegrated atoms pass, there is in most cases a marked alteration in the chemical and physical properties of the matter. 499The transformation of the radio-elements is thus a transformation of a part per saltum, and not a progressive change of the whole. At any time after the process of transformation has been in progress there will thus remain a part of the matter which is unchanged, and, mixed with it, the products which have resulted from the transformation of the remainder.
The question naturally arises whether the process of degradation of matter is confined to the radio-elements or is a universal property of matter. It will be shown in chapter XIV that all matter, so far examined, exhibits the property of radio-activity to a slight degree. It is very difficult, however, to make certain that the observed radio-activity is not due to the presence in the matter of a slight trace of a radio-element. If ordinary matter is radio-active, it is certain that its activity is much less than that of uranium, and consequently that its rate of transformation must be excessively slow. There is, however, another possibility to be considered. The changes occurring in the radio-elements would probably never have been detected if the change had not been accompanied by the expulsion of charged particles with great velocity. It does not seem unlikely that an atom may undergo disintegration without projecting a part of its system with sufficient velocity to ionize the gas. In fact, we have seen that, even in the radio-elements, several of the series of changes in both thorium, radium, and actinium are unaccompanied by ionizing rays. The experimental results given in Appendix A strongly support this point of view. It may thus be possible that all matter is undergoing a slow process of transformation, which has so far only been detected in the radio-elements on account of the expulsion of charged particles with great velocity during the change. This process of degradation of matter continuing for ages must reduce the constituents of the earth to the simpler and more stable forms of matter.
The idea that helium is a transformation product of radium suggests the probability that helium is one of the more elementary substances of which the heavier atoms are composed. Sir Norman Lockyer, in his interesting book on “Inorganic Evolution,” has pointed out that the spectra of helium and of hydrogen predominate in the hottest stars. In the cooler stars the more 500complex types of matter appear. Sir Norman Lockyer has based his theory of evolution of matter on evidence of a spectroscopic examination of the stars, and considers that temperature is the main factor in breaking up matter into its simpler forms. The transformation of matter occurring in the radio-elements is on the other hand spontaneous, and independent of temperature over the range examined.
273. Radio-activity of the atmosphere. The experiments of Geitel[384] and C. T. R. Wilson[385] in 1900 showed that a positively or negatively charged conductor placed inside a closed vessel gradually lost its charge. This loss of charge was shown to be due to a small ionization of the air inside the vessel. Elster and Geitel also found that a charged body exposed in the open air lost its charge rapidly, and that the rate of discharge was dependent on the locality and on atmospheric conditions. A more detailed description and discussion of these results will be given later in section 284.
In the course of these experiments, Geitel observed that the rate of discharge increased slightly for some time after the vessel had been closed. He considered that this might possibly be due to the existence of some radio-active substances in the air, which produced excited activity on the walls of the vessel and so increased the rate of dissipation of the charge. In 1901 Elster and Geitel[386] tried the bold experiment of seeing whether it were possible to extract a radio-active substance from the air. The experiments of the writer had shown that the excited radio-activity from the thorium emanation could be concentrated on the negative electrode in a strong electric field. This result indicated that the carriers of the radio-activity had a positive charge of 502electricity. Elster and Geitel therefore tried an experiment to see whether positively charged carriers, possessing a similar property, were present in the atmosphere. For this purpose a cylinder of wire-netting, charged negatively to 600 volts, was exposed for several hours in the open air. The cylinder was then removed, and quickly placed in a large bell-jar, inside which was placed an electroscope to detect the rate of discharge. It was found that the rate of discharge was increased to a slight extent. In order to multiply the effect a wire about 20 metres in length was exposed at some height from the ground, and was kept charged to a high potential by connecting it to the negative terminal of an influence machine. After exposure for some hours, this wire was removed and placed inside the dissipation vessel. The rate of discharge was found to be increased many times by the presence of the wire. No increase was observed when the wire was charged positively instead of negatively. The results also showed that the radio-active matter could be removed from the wire in the same way as from a wire made active by exposure in the presence of the thorium emanation. A piece of leather moistened with ammonia was rubbed over the active wire. On testing the leather, it was found to be strongly radio-active. When a long wire was used, the amount of activity obtained on the leather was comparable with that possessed by a gram of uranium oxide.
The activity produced on the wire was not permanent, but disappeared to a large extent in the course of a few hours. The amount of activity produced on a wire of given size, exposed under similar conditions, was independent of the material of the wire. Lead, iron and copper wires gave about equal effects.
The amount of activity obtained was greatly increased by exposing a negatively charged wire in a mass of air which had been undisturbed for a long time. Experiments were made in the great cave of Wolfenbüttel, and a very large amount of activity was observed. By transferring the activity to a piece of leather it was found that the rays could appreciably light up a screen of barium platinocyanide in the dark[387]. The rays also darkened a photographic plate through a piece of aluminium 0·1 mm. in thickness.
503These remarkable experiments show that the excited radio-activity obtained from the atmosphere is very similar in character to the excited activity produced by the emanations of radium and thorium. No investigators have contributed more to our knowledge of the radio-activity and ionization of the atmosphere than Elster and Geitel. The experiments here described have been the starting-point of a series of researches by them and others on the radio-active properties of the atmosphere, which have led to a great extension of our knowledge of that important subject.
Rutherford and Allan[388] determined the rate of decay of the excited activity produced on a negatively charged wire exposed in the open air. A wire about 15 metres long was exposed in the open air, and kept charged by an influence machine to a potential of about -10,000 volts. An hour’s exposure was sufficient to obtain a large amount of excited activity on the wire. The wire was then rapidly removed and wound on a framework which formed the central electrode in a large cylindrical metal vessel. The ionization current for a saturation voltage was measured by means of a sensitive Dolezalek electrometer. The current, which is a measure of the activity of the wire, was found to diminish according to an exponential law with the time, falling to half value in about 45 minutes. The rate of decay was independent of the material of the wire, of the time of exposure, and of the potential of the wire.
An examination was also made of the nature of the rays emitted by the radio-active wire. For this purpose a lead wire was made radio-active in the manner described, and then rapidly wound into the form of a flat spiral. The penetrating power of the rays was tested in a vessel similar to that shown in Fig. 17. Most of the ionization was found to be due to some very easily absorbed rays, which were of a slightly more penetrating character than the α rays emitted from a wire made active by the radium or thorium emanations. The intensity of the rays was cut down to half value by about 0·001 cm. of aluminium. The photographic action observed by Elster and Geitel through 0·1 mm. of aluminium showed that some penetrating rays were also present. This was afterwards confirmed by Allan, who used the electric method. These penetrating 504rays are probably similar in character to the β rays from the radio-elements.
274. The excited activity produced on the negatively charged wire cannot be due to an action of the strong electric field on the surface of the wire; for very little excited activity is produced if the wire is charged to the same potential inside a closed cylinder.
We have seen that the excited activity produced on the wire can be partially removed by rubbing and by solution in acids, and, in this respect, it is similar to the excited activity produced in bodies by the emanations of radium and thorium. The very close similarity of the excited activity obtained from the atmosphere to that obtained from the radium and thorium emanations suggests the probability that a radio-active emanation exists in the atmosphere. This view is confirmed by a large amount of indirect evidence discussed in sections 276, 277 and 280.
Assuming the presence of a radio-active emanation in the atmosphere, the radio-active effects observed receive a simple explanation. The emanation in the air gradually breaks up, giving rise in some way to positively charged radio-active carriers. These are driven to the negative electrode in the electric field, and there undergo a further change, giving rise to the radiations observed at the surface of the wire. The matter which causes excited activity will thus be analogous to the active deposit of radium and thorium.
Since the earth is negatively electrified with regard to the upper atmosphere, these positive radio-active carriers produced in the air are continuously deposited on the surface of the earth. Everything on the surface of the earth, including the external surface of buildings, the grass, and leaves of trees, must be covered with an invisible deposit of radio-active material. A hill, or mountain peak, or any high mass of rock or land, concentrates the earth’s electric field at that point and consequently will receive more excited radio-activity per unit area than the plain. Elster and Geitel have pointed out that the greater ionization of the air observed in the neighbourhood of projecting peaks receives a satisfactory explanation on this view.
If the radio-active carriers are produced at a uniform rate in 505the atmosphere, the amount of excited activity It, produced on a wire exposed under given conditions, will, after exposure for a time t, be given by
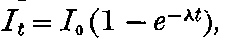
where I₀ is the maximum activity on the wire and λ is the constant of decay of the excited activity. Since the activity of a wire after removal falls to half value in about 45 minutes, the value of λ is 0·92 (hour)-1. Some experiments made by Allan[389] are in rough agreement with the above equation. Accurate comparative results are difficult to obtain on account of the inconstancy of the radio-activity of the open air. After an exposure of a wire for several hours, the activity reached a practical maximum, and was not much increased by continued exposure.
We have seen (section 191) that the carriers of the active deposit of radium and thorium move in an electric field with about the same velocity as the ions. We should expect therefore that a long wire charged to a high negative potential would abstract the active carriers from the atmosphere for a considerable distance. This does not appear to be the case, for Eve (see section 281) has found that the carriers are only abstracted from the air for a radius of less than one metre, for a potential of the wire of -10,000 volts. It seems probable that the carriers of the active matter are deposited on the numerous fine dust particles present in the air and thus move very slowly even in a strong electric field.
The amount of excited activity produced on a wire, supported some distance from the surface of the earth, should increase steadily with the voltage, for the greater the potential, the greater the volume of air from which the radio-active carriers are abstracted.
The presence of radio-active matter in the atmosphere will account for a considerable portion of the ionization of the air observed near the earth. This important question is discussed in more detail in section 281.
275. Radio-activity of freshly fallen rain and snow. C. T. R. Wilson[390] tried experiments to see if any of the radio-active material from the air was carried down by rain. For this purpose a quantity of freshly fallen rain was collected, rapidly 506evaporated to dryness in a platinum vessel, and the activity of the residue tested by placing the vessel in an electroscope. In all cases, the rate of discharge of the electroscope was considerably increased. From about 50 c.c. of rain water, an amount of activity was obtained sufficient to increase the rate of discharge of the electroscope four or five times, after the rays had traversed a thin layer of aluminium or gold-leaf. The activity disappeared in the course of a few hours, falling to half value in about 30 minutes. Rain water, which had stood for some hours, showed no trace of activity. Tap water, when evaporated, left no active residue.
The amounts of activity obtained from a given quantity of rain water were all of the same order of magnitude, whether the rain was precipitated in fine or in large drops, by night or by day, or whether the rain was tested at the beginning or at the end of a heavy rainfall lasting several hours.
The activity obtained from rain is not destroyed by heating the platinum vessel to a red heat. In this and other respects it resembles the excited activity obtained on negatively charged wires exposed in the open air.
C. T. R. Wilson[391] obtained a radio-active precipitate from rain water by adding a little barium chloride and precipitating the barium with sulphuric acid. An active precipitate was also obtained when alum was added to the water, and the aluminium precipitated by ammonia. The precipitates obtained in this way showed a large activity. The filtrate when boiled down was quite inactive, showing that the active matter had been completely removed by precipitation. This effect is quite analogous to the production of active precipitates from a solution containing the active deposit of thorium (see section 185).
The radio-activity of freshly fallen snow was independently observed by C. T. R. Wilson[392] in England, and Allan[393] and McLennan[394] in Canada. In order to obtain a large amount of activity, the surface layer of snow was removed, and evaporated to dryness in a metal vessel. An active residue was obtained with radio-active 507properties similar to those observed for freshly fallen rain. Both Wilson and Allan found that the activity of rain and snow decayed at about the same rate, the activity falling to half value in about 30 minutes. McLennan states that he found a smaller amount of radio-activity in the air after a prolonged fall of snow.
Schmauss[395] has observed that drops of water falling through air ionized by Röntgen rays acquire a negative charge. This effect is ascribed to the fact that the negative ions in air diffuse faster than the positive. On this view the drops of rain and flakes of snow would acquire a negative charge in falling through the air. They would in consequence act as collectors of the positive radio-active carriers from the air. On evaporation of the water the radio-active matter would be left behind.
276. Radio-active emanations from the earth. Elster and Geitel observed that the air in caves and cellars was, in most cases, abnormally radio-active, and showed very strong ionization. This action might possibly be due to an effect of stagnant air, by which it produced a radio-active emanation from itself, or to a diffusion of a radio-active emanation from the soil. To test whether this emanation was produced by the air itself, Elster and Geitel shut up the air for several weeks in a large boiler, but no appreciable increase of the activity or ionization was observed. To see whether the air imprisoned in the capillaries of the soil was radio-active, Elster and Geitel[396] put a pipe into the earth and sucked up the air into a testing vessel by means of a water pump.
The apparatus employed to test the ionization of the air is shown in Fig. 103. C is an electroscope connected with a wire net, Z. The active air was introduced into a large bell-jar of 27 litres capacity, the inside of which was covered with wire netting, MM´. The bell-jar rested on an iron plate AB. The electroscope could be charged by the rod S. The rate of discharge of the electroscope, before the active air was introduced, was noted. On allowing the active air to enter, the rate of discharge increased rapidly, rising in the course of a few hours in one experiment to 30 times the original value. They found that the emanation produced 508excited activity on the walls of the containing vessel. The air sucked up from the earth was even more active than that observed in caves and cellars. There can thus be little doubt that the abnormal activity observed in caves and cellars is due to a radio-active emanation, present in the earth, which gradually diffuses to the surface and collects in places where the air is not disturbed.
Results similar to those obtained by Elster and Geitel for the air removed from the earth at Wolfenbüttel were also obtained later by Ebert and Ewers[397] at Munich. They found a strongly active emanation in the soil, and, in addition, examined the variation with time of the activity due to the emanation in a sealed vessel. After the introduction of the active air into the testing vessel, the activity was observed to increase for several hours, and then to decay, according to an exponential law, with the time, falling to half value in about 3·2 days. This rate of decay is more rapid than that observed for the radium emanation, which decays to half value in a little less than four days. The increase of activity with time is probably due to the production of excited activity on the walls of the vessel by the emanation. In this respect it is analogous to the increase of activity observed when the radium emanation is introduced into a closed vessel. No definite experiments were made by Ebert and Ewers on the rate of decay of this excited activity. In one experiment the active emanation, after standing in the vessel for 140 hours, was removed by sucking ordinary air of small activity through the apparatus. The activity rapidly fell to about half value, and this was followed by a very slow decrease of the activity with time. This result indicates that about half the rate of discharge observed was due to the radiation from the emanation and the other half to the excited activity produced by it.
The apparatus employed by Ebert and Ewers in these experiments was very similar to that employed by Elster and Geitel, shown in Fig. 103. Ebert and Ewers observed that, when the wire net attached to the electroscope was charged negatively, the rate of discharge observed was always greater than when it was charged positively. The differences observed between the two rates of discharge varied between 10 and 20 per cent. A similar effect 509has been observed by Sarasin, Tommasina and Micheli[398] for a wire made active by exposure to the open air. This difference in the rates of discharge for positive and negative electricity is probably connected with the presence of particles of dust or small water globules suspended in the gas. The experiments of Miss Brooks (section 181) have shown that the particles of dust present in the air containing the thorium emanation become radio-active. A large proportion of these dust particles acquire a positive charge and are carried to the negative electrode in an electric field. This effect would increase the rate of discharge of the electroscope when charged negatively. In later experiments, Ebert and Ewers noticed that, in some cases, when the air had been kept in the vessel for several days, the effect was reversed, and the electroscope showed a great rate of discharge when charged positively.
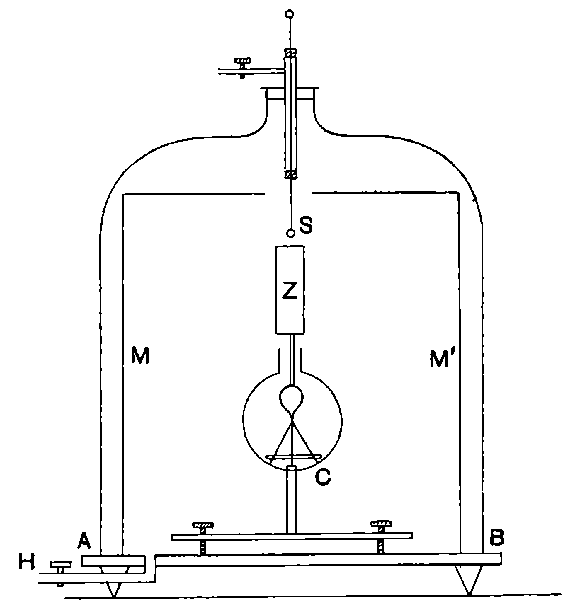
Fig. 103.
J. J. Thomson[399] has observed that the magnitude of the ionization current depends on the direction of the electric field, if fine water globules are suspended in the ionized gas.
510In later experiments, Ebert[400] found that the radio-active emanation could be removed from the air by condensation in liquid air. This property of the emanation was independently discovered by Ebert before he was aware of the results of Rutherford and Soddy on the condensation of the emanations of radium and thorium. To increase the amount of radio-active emanation in a given volume of air, a quantity of the active air, obtained by sucking the air from the soil, was condensed by a liquid air machine. The air was then allowed partially to evaporate, but the process was stopped before the point of volatilization of the emanation was reached. This process was repeated with another quantity of air and the residues added together. Proceeding in this way, he was able to concentrate the emanation in a small volume of air. On allowing the air to evaporate, the ionization of the air in the testing vessel increased rapidly for a time and then slowly diminished. Ebert states that the maximum for the emanation which had been liquefied for some time was reached earlier than for fresh air. The rate of decay of activity of the emanation was not altered by keeping it at the temperature of liquid air for some time. In this respect it behaves like the emanations of radium and thorium.
J. J. Thomson[401] found that air bubbled through Cambridge tap water showed much greater conductivity than ordinary air. The air was drawn through the water by means of a water pump into a large gasometer, when the ionization current was tested with a sensitive electrometer. When a rod charged negatively was introduced into this conducting air it became active. After an exposure for a period of 15 to 30 minutes in the conducting gas, the rod, when introduced into a second testing vessel, increased the saturation current in the vessel to about five times the normal amount. Very little effect was produced when the rod was uncharged or charged positively for the same time. The activity of the rod decayed with the time, falling to half value in about 40 minutes. The amount of activity produced on a wire under constant conditions was independent of the material of the wire. The rays from the rod were readily absorbed in a few centimetres of air.
These effects were, at first, thought to be due to the action of 511the small water drops suspended in the gas, for it was well known that air rapidly drawn through water causes a temporary increase in its conductivity. Later results, however, showed that there was a radio-active emanation present in Cambridge tap water. This led to an examination of the waters from deep wells in various parts of England, and J. J. Thomson found that, in some cases, a large amount of emanation could be obtained from the well water. The emanation was released either by bubbling air through the water or by boiling the water. The gases obtained by boiling the water were found to be strongly active. A sample of air mixed with the radio-active emanation was condensed. The liquefied gas was allowed to evaporate, and the earlier and later portions of the gas were collected in separate vessels. The final portion was found to be about 30 times as active as the first portion.
An examination of the radio-active properties of the active gases so obtained has been made by Adams[402]. He found that the activity of the emanation decayed, according to an exponential law, with the time, falling to half value in about 3·4 days. This is not very different from the rate of decay of the activity of the radium emanation, which falls to half value in a little less than four days. The excited activity produced by the emanation decayed to half value in about 35 minutes. The decay of the excited activity from radium is at first irregular, but after some time falls off, according to an exponential law, diminishing to half value in 28 minutes. Taking into account the uncertainty attaching to measurements of the very small ionization observed in these experiments, the results indicate that the emanation obtained from well water in England is similar to, if not identical with, the radium emanation. Adams observed that the emanation was slightly soluble in water. After well water had been boiled for a while and then put aside, it was found to recover its power of giving off an emanation. The amount obtained after standing for some time was never more than 10 per cent. of the amount first obtained. Thus it is probable that the well water, in addition to the emanations mixed with it, has also a slight amount of a permanent radio-active substance dissolved in it. Ordinary rain water or distilled water does not give off an emanation.
512Bumstead and Wheeler[403] have made a very careful examination of the radio-activity of the emanation obtained from the surface water and soil at New Haven, Connecticut. The emanation, obtained from the water by boiling, was passed into a large testing cylinder, and measurements of the current were made by means of a sensitive electrometer. The current gradually rose to a maximum, after the introduction of the emanation, in exactly the same way as the current increases in a vessel after the introduction of the radium emanation. The decay of activity of the emanations obtained from the water and soil was carefully measured, and, within the limits of experimental error, agreed with the rate of decay of activity observed for the radium emanation. The identity of the emanations from the water and soil with the radium emanation was still further established by experiments on the rate of diffusion of the emanation through a porous plate. By comparative tests it was found that the coefficient of diffusion of the emanations from the water and soil was the same as for the radium emanation. Also, by comparison of the rate of diffusion of carbonic acid, it was found that the density of the emanation was about four times that of carbonic acid, a result in good agreement with that found for the radium emanation (sections 161 and 162).
Bumstead[404] has found that a considerable amount of thorium as well as radium emanation exists in the air of New Haven. For a three hour exposure in the open air, 3 to 5 per cent. of the excited activity on the wire is due to thorium. For a twelve hour exposure, the thorium activity was sometimes 15 per cent. of the whole. On account of the comparatively slow decay of the excited activity of thorium, the activity on the wire after removal for three or four hours was due almost entirely to thorium. The rate of decay could then be measured accurately, and was found to be the same as for a wire exposed in the presence of the thorium emanation.
Dadourian[405] has made an examination of the underground air in New Haven, and has found that this too contains a large 513quantity of the thorium emanation. A circular hole about 50 cms. in diameter and 2 metres deep was dug in the ground. A number of wires were wound on an insulated frame and suspended in the hole, the top of the hole then being covered over. The wire was charged negatively by a Wimshurst machine. After a long exposure the excited activity on the wire diminished at a rate that showed it to be a mixture of the excited activities of thorium and radium.
A very large amount of work has been done in examining various hot and mineral springs for the presence of the radium emanation, and it is not possible here to refer more than briefly to a few of the very numerous papers that have been published on this subject both in Europe and America. H. S. Allen and Lord Blythswood[406] have observed that the hot springs at Bath and Buxton gave off a radio-active emanation. This was confirmed by Strutt[407], who found that the escaping gases contained the radium emanation, and also that the mud deposited from the springs contained a trace of radium salts. These results are of considerable interest, for Lord Rayleigh has observed that helium is contained among the gases evolved by the springs. It appears probable that the helium observed is produced from the radium or radio-active deposits through which the water flows. Many mineral and hot springs which are famous for their curative properties have been found to contain traces of radium and also considerable amounts of radium emanation. It has been suggested that the curative properties may be due to some extent to the presence of these minute quantities of radium.
Himstedt[408] found that the thermal springs at Baden Baden contained the radium emanation, while Elster and Geitel[409] examined the deposits formed by these springs and found them to contain small quantities of radium salts. Results of a similar character were obtained for a number of waters in Germany by Dorn[410], Schenck[411], and H. Mache[412].
514Curie and Laborde[413] have tested the waters of a large number of mineral springs and found that the great majority contain the radium emanation. In this connection, it is of interest to note that Curie and Laborde found very little emanation in the waters of Salins-Moutiers, while Blanc[414] observed, on the other hand, that the sediment from the spring was very active. A closer examination of this deposit by Blanc revealed the fact that it contained a considerable quantity of thorium. This was proved by finding that it gave out an emanation, which lost half of its activity in one minute, and produced excited activity, which fell to half value in about 11 hours. Boltwood[415] has tested a number of samples of spring water from different sources in America and has found that many of them contain the radium emanation.
Most of the results upon the amount of radium emanation from different sources have been expressed in arbitrary units without, in many cases, any comparative standard being given. Boltwood (loc. cit.) has described a satisfactory method for collecting and testing the emanation from different waters, and has suggested that the rate of discharge observed by the electroscope or the electrometer should be expressed in terms of the effect due to the emanation liberated on solution of a definite weight of the mineral uraninite. Since in every mineral so far examined, the amount of radium present is proportional to the amount of uranium, such a standard would be sufficiently definite for practical purposes. The emanation liberated from a few centigrams of the mineral is sufficient to give a convenient rate of discharge of an electroscope. Such a method is preferable to using a known quantity of a radium compound as a standard, since it is difficult to know with certainty the activity of the preparations of radium which may be in the possession of the different experimenters.
277. Radio-activity of constituents of the earth. Elster and Geitel[416] observed that, although in many cases the conductivity of the air was abnormally high in underground enclosures, the conductivity varied greatly in different places. In the Baumann 515Cave, for example, the conductivity of the air was nine times the normal, but in the Iberg Cave only three times the normal. In a cellar at Clausthal the conductivity was only slightly greater than the normal, but the excited radio-activity obtained on a negatively charged wire exposed in it was only ¹⁄₁₁ of the excited radio-activity obtained when the wire was exposed in the free air. They concluded from these experiments that the amount of radio-activity in the different places probably varied with the nature of the soil. Observations were then made on the conductivity of the air sucked up from the earth at different parts of the country. The clayey and limestone soils at Wolfenbüttel were found to be strongly active, the conductivity varying from four to sixteen times the normal amount. A sample of air from the shell limestone of Würzburg and from the basalt of Wilhelmshöhe showed very little activity.
Experiments were made to see whether any radio-active substance could be detected in the soil itself. For this purpose some earth was placed on a dish and introduced under a bell-jar, similar to that shown in Fig. 103. The conductivity of the air in the bell-jar increased with the time, rising to three times the normal value after several days. Little difference was observed whether the earth was dry or moist. The activity of the soil seemed to be permanent, for no change in the activity was observed after the earth had been laid aside for eight months.
Attempts were then made to separate the radio-active constituent from the soil by chemical treatment. For this purpose a sample of clay was tested. By extraction with hydrochloric acid all the calcium carbonate was removed. On drying the clay the activity was found to be reduced, but it spontaneously regained its original activity in the course of a few days. It seems probable, therefore, that an active product had been separated from the soil by the acid. Elster and Geitel consider that an active substance was present in the clay, which formed a product more readily soluble in hydrochloric acid than the active material itself. There seemed to be a process of separation analogous to that of Th X from thorium by precipitation with ammonia.
Experiments were also made to see whether substances placed in the earth acquired any radio-activity. For this purpose samples 516of potter’s clay, whitening, and heavy spar, wrapped in linen, were placed in the earth 50 cms. below the surface. After an interval of a month, these were dug up and their activity examined. The clay was the only substance which showed any activity. The activity of the clay diminished with the time, showing that activity had been excited in it by the emanations present in the soil.
Elster and Geitel[417] have found that a large quantity of the radio-active emanation can be obtained by sucking air through clay. In some cases, the conductivity of the air in the testing vessel was increased over 100 times. They have also found that the so-called “fango”—a fine mud obtained from hot springs in Battaglia, Northern Italy—gives off three or four times as much emanation as clay. By treating the fango with acid, the active substance present was dissolved. On adding some barium chloride to the solution, and precipitating the barium as sulphate, the active substance was removed, and in this way a precipitate was obtained over 100 times as active, weight for weight, as the original fango. Comparisons were made of the rate of decay of the excited activity, due to the emanation from fango, with that due to the radium emanation, and within the limits of error, the decay curves obtained were found to be identical. There can thus be no doubt that the activity observed in fango is due to the presence of a small quantity of radium. Elster and Geitel calculate that the amount of radium, contained in it, is only about one-thousandth of the amount to be obtained from an equal weight of pitchblende from Joachimsthal.
Vincenti and Levi Da Zara[418] have found that the waters and sediments of a number of hot springs in Northern Italy contain the radium emanation. Elster and Geitel observed that natural carbonic acid obtained from great depths of old volcanic soil was radio-active, while Burton[419] found that the petroleum from a deep well in Ontario, Canada, contained a large quantity of emanation, probably of radium, since its activity fell to half value in 3·1 days, while the excited activity produced by the emanation fell to half 517value in about 35 minutes. A permanently active deposit was left behind after volatilization of the oil, indicating that probably one or more of the radio-elements were present in minute quantity.
Elster and Geitel[420] have found that the active sediments obtained from springs at Nauheim and Baden Baden showed abnormal rates of decay of the excited activity. This was finally traced to the presence in the deposit of both thorium and radium. By suitable chemical methods, the two active substances were separated from each other and were then tested separately.
278. Effect of meteorological conditions upon the radio-activity of the atmosphere. The original experiments of Elster and Geitel on the excited radio-activity derived from the atmosphere were repeated by Rutherford and Allan[421] in Canada. It was found that a large amount of excited radio-activity could be derived from the air, and that the effects were similar to those observed by Elster and Geitel in Germany. This was the case even on the coldest day in winter, when the ground was covered deeply with snow and wind was blowing from the north over snow-covered lands. The results showed that the radio-activity present in the air was not much affected by the presence of moisture, for the air during a Canadian winter is extremely dry. The greatest amount of excited activity on a negatively charged wire was obtained in a strong wind. In some cases the amount produced for a given time of exposure was ten to twenty times the normal amount. A cold bright day of winter usually gave more effect than a warm dull day in summer.
Elster and Geitel[422] have made a detailed examination of the effect of meteorological conditions on the amount of excited radio-activity to be derived from the atmosphere. For this purpose a simple portable apparatus was devised by them and used for the whole series of experiments. A large number of observations were taken, extending over a period of twelve months. They found that the amount of excited activity obtained was subject to great 518variations. The extreme values obtained varied in the ratio of 16 to 1. No direct connection could be traced between the amount of ionization in the atmosphere and the amount of excited activity produced. They found that the greatest amount of excited activity was obtained during a fog, when the amount of ionization in the air was small. This result, however, is not necessarily contradictory to the view that the ionization and activity of the air are to a certain extent connected. From the experiments of Miss Brooks on the effect of dust in acting as carriers of excited activity, more excited activity should be obtained during a fog than in clear air. The particles of water become centres for the deposit of radio-active matter. The positive carriers are thus anchored and are not removed from the air by the earth’s field. In a strong electric field, these small drops will be carried to the negative electrode and manifest their activity on the surface of the wire. On the other hand, the distribution of water globules throughout the air causes the ions in the air to disappear rapidly in consequence of their diffusion to the surface of the drops (see section 31). For this reason the denser the fog, the smaller will be the conductivity observed in the air.
Lowering the temperature of the air had a decided influence. The average activity observed below 0° C. was 1·44 times the activity observed above 0° C. The height of the barometer was found to exert a marked influence on the amount of excited activity to be derived from the air. The lower the barometer the greater was the amount of excited activity in the air. The effect of variation of the height of the barometer is intelligible, when it is considered that probably a large proportion of the radio-activity observed in the air is due to the radio-active emanations which are continuously diffusing from the earth into the atmosphere. Elster and Geitel have suggested that a lowering of the pressure of the air would cause the air from the ground to be drawn up from the capillaries of the earth into the atmosphere. This, however, need not necessarily be the case if the conditions of the escape of the emanation into the atmosphere are altered by the variation of the position of underground water or by a heavy fall of rain.
The amount of excited activity to be derived from the air on the Baltic Coast was only one-third of that observed inland at 519Wolfenbüttel. Experiments on the radio-activity of the air in mid-ocean would be of great importance in order to settle whether the radio-activity observed in the air is due to the emanations from the soil alone. It is probable that the radio-activity of the air at different points of the earth may vary widely, and may largely depend on the nature of the soil.
Saake[423] has found that the amount of emanation present in the air at high altitudes in the valley of Arosa in Switzerland is much greater than the normal amount at lower levels. Elster and Geitel have observed that there is also a larger number of ions in the air at high altitudes, and suggest that the curative effect of thermal springs and the physiological actions of the air at high levels may be connected with the presence of an unusual amount of radio-active matter in the atmosphere. Simpson[424] made experiments on the amount of excited activity at Karasjoh, Norway, at a height of about 150 feet above sea level. The sun did not rise above the level of the horizon during the time the observations were taken. The average amount of excited activity obtained from the air was considerably greater than the normal amount observed by Elster and Geitel in Germany. This was the more surprising as the ground was frozen hard and covered with deep snow. Allan, working in Montreal, Canada, early observed that the amount of activity to be obtained from the air was about the same in summer as in winter, although, in the latter case, the whole earth was deeply frozen and covered with snow, and the winds blew from the north over snow-covered lands. Under such conditions, a diminution of the amount of activity is to be expected since the diffusion of the emanation must be retarded, if not altogether stopped, by the freezing of the soil. On the other hand, it appears difficult to escape from the conclusion of Elster and Geitel that the emanation present in the atmosphere is evolved from the earth itself.
Some interesting experiments have been made by McLennan[425] on the amount of excited radio-activity to be derived from the air when filled with fine spray. The experiments were made at the 520foot of the American Fall at Niagara. An insulated wire was suspended near the foot of the Fall, and the amount of excited activity on the wire compared with the amount to be obtained on the same wire for the same exposure in Toronto. The amount of activity obtained from the air at Toronto was generally five or six times that obtained from the air at the Falls. In these experiments it was not necessary to use an electric machine to charge the wire negatively, for the falling spray kept the insulated wire permanently charged to a potential of about -7500 volts. These results indicate that the falling spray had a negative charge and electrified the wire. The small amount of the excited radio-activity at the Falls was probably due to the fact that the negatively charged drops abstracted the positively charged radio-active carriers from the atmosphere, and in falling carried them to the river below. On collecting the spray and evaporating it, no active residue was obtained. Such a result is, however, to be expected on account of the minute proportion of the spray tested compared with that present in the air.
279. A very penetrating radiation from the earth’s surface. McLennan[426], and Rutherford and Cooke[427] independently, observed the presence of a very penetrating radiation inside buildings. McLennan measured the natural conductivity of the air in a large closed metal cylinder by means of a sensitive electrometer. The cylinder was then placed inside another and the space between filled with water. For a thickness of water between the cylinders of 25 cms. the conductivity of the air in the inner cylinder fell to about 63 per cent. of its initial value. This result shows that part of the ionization in the inner cylinder was due to a penetrating radiation from an external source, which radiation was partially or wholly absorbed in water.
Rutherford and Cooke observed that the rate of discharge of a sealed brass electroscope was diminished by placing a lead screen around the electroscope. A detailed investigation of the decrease of the rate of discharge in the electroscope, when surrounded by metal screens, was made later by Cooke[428]. A thickness of 5 cms. of lead 521round the electroscope decreased the rate of discharge about 30 per cent. Further increase of the thickness of the screen had no effect. When the apparatus was surrounded by 5 tons of pig-lead the rate of discharge was about the same as when it was surrounded by a plate about 3 cms. thick. An iron screen also diminished the rate of discharge to about the same extent as the lead. By suitably arranging lead screens it was found that the radiation came equally from all directions. It was of the same intensity by night as by day. In order to be sure that this penetrating radiation did not arise from the presence of radio-active substances in the laboratory, the experiments were repeated in buildings in which radio-active substances had never been introduced, and also on the open ground far removed from any building. In all cases a diminution of the rate of discharge of the electroscope, when surrounded by lead screens, was observed. These results show that a penetrating radiation is present at the surface of the earth, arising partly from the earth itself and partly from the atmosphere.
The result is not surprising when the radio-activity of the earth and atmosphere is taken into account. The writer has found that bodies made active by exposure to the emanations from thorium and radium give out γ rays. We may expect then that the very similar excited radio-activity which is present in the earth and atmosphere should also give rise to γ rays of a similar character. More recent work, however (section 286), indicates that this explanation is not sufficient to explain all the facts observed.
280. Comparison of the radio-activity of the atmosphere with that produced by the radio-elements. The radio-active phenomena observed in the earth and atmosphere are very similar in character to those produced by thorium and radium. Radio-active emanations are present in the air of caves and cellars, in natural carbonic acid, and in deep well water, and these emanations produce excited radio-activity on all bodies in contact with them. The question now arises whether these effects are due entirely to known radio-elements present in the earth or to unknown kinds of radio-active matter. The simplest method of testing this point is to compare the rate of decay of the radio-active 522product in the atmosphere with those of the known radio-active products of thorium and radium. A cursory examination of the facts at once shows that the radio-activity of the atmosphere is much more closely allied to effects produced by radium than to those due to thorium. The activity of the emanation released from well water, and also that sucked up from the earth, decays to half value in about 3·3 days, while the activity of the radium emanation decays to half value in an interval of 3·7 to 4 days. Considering the difficulty of making accurate determinations of these quantities, the rates of decay of the activity of the emanations from the earth and from radium agree within the limits of experimental error. A large number of observers have found that the radium emanation is present in the water of thermal springs and in the sediment deposited by them. Bumstead and Wheeler have shown that the emanation from the soil and surface water of New Haven is identical with that from radium. If the emanations from the earth and from radium are the same, the excited activities produced should have the same rate of decay. The emanation from well water in England approximately fulfils this condition (section 276), but an observation recorded by Ebert and Ewers (section 276) seems to show that the excited activity due to the emanation sucked up from the earth decays at a very slow rate compared with that due to radium.
Bumstead has given undoubted evidence that the thorium as well as the radium emanation is also present in the atmosphere at New Haven, while Dadourian has shown that it is emitted by New Haven soil. Blanc, and Elster and Geitel, have also found that thorium is present in the sediment from some thermal springs.
If the active matter in the atmosphere consists mainly of the radium emanation, the active deposit on a negatively charged wire, exposed in the open air, should initially consist of radium A, B and C. The curve of decay should be identical with the decay curve of the excited activity of radium, measured by the α rays, that is, there should be a rapid initial drop corresponding to the initial 3 minute change, then a slow rate of variation, the activity after several hours decaying to half value in about 28 minutes (see section 222). The rapid initial drop has been observed by 523Bumstead for the air at New Haven. Allan[429] did not observe this initial drop in Montreal, but found the activity fell to half value in about 45 minutes, reckoning from a time about 10 minutes after the removal of the active wire. This is about the rate of decay to be expected for the active deposit of radium over the same interval. Allan obtained evidence that there were several kinds of active matter deposited on the wire. For example, the activity transferred from the active wire to a piece of leather, moistened with ammonia, fell to half value in 38 minutes; for a piece of absorbent felt treated similarly, the activity fell to half value in 60 minutes, the normal time for the untreated wire being 45 minutes.
It is probable that this variation of the rate of decay is due to the fact that unequal proportions of radium B and C were transferred from the wire to the rubber. If a greater proportion of B than of C were removed, the decay would be slower and vice versa.
The fact that the activity of rain and snow falls to half value in about 30 minutes is a strong indication that the radium emanation is present in the atmosphere. The active matter with the rain and snow after standing some time would consist mainly of radium C and this should decay exponentially with the time, falling to half value in 28 minutes.
On account of the rapid decay of the thorium emanation—half value in one minute—it is not likely that much of the activity of the atmosphere can be ascribed to it. Its effect would be most marked near the surface of the soil.
There can be little doubt, that a large part of the radio-activity of the atmosphere is due to the radium emanation, which is continually diffusing into the atmosphere from the pores of the earth. Since radio-activity has been observed in the atmosphere at all points at which observations have, so far, been made, radio-active matter must be distributed in minute quantities throughout the soil of the earth. The volatile emanations escape into the atmosphere by diffusion, or are carried to the surface in spring water or by the escape of underground gases, and cause the radio-active phenomena observed in the atmosphere. The observation of Elster and Geitel that the radio-activity of the air is much less near the sea than inland is explained at once, if the radio-activity of the 524atmosphere is due mainly to the diffusion of emanations from the soil into the air above it.
The rare gases helium and xenon which exist in the atmosphere have been tested and found to be non-radio-active. The radio-activity of the air cannot be ascribed to a slight radio-activity possessed by either of these gases.
281. Amount of the radium emanation in the atmosphere. It is a matter of great interest to form an estimate of the amount of radium emanation present in the atmosphere, for since it comes from the earth, it indirectly serves as a means of estimating the amount of radium which is distributed over a thin crust of the earth.
Some experiments in this direction have been made by Eve in the laboratory of the writer. The experiments are not yet completed but the results so far obtained allow us to calculate the probable amount of emanation per cubic kilometre of the atmosphere near the earth.
Experiments were first made with a large iron tank 154 cms. square and 730 cms. deep, in a building in which no radium or other radio-active material had ever been introduced. The saturation ionization current for the air in the tank was first measured by means of an electroscope, connected with an insulated electrode passing up the centre of the closed tank. Assuming that the ionization in the tank was uniform, the number of ions produced per c.c. of the air in the tank was found to be 10. This is a considerably lower value than has usually been observed in a small closed vessel (see section 284). Cooke obtained the value 10 for a well cleaned brass electroscope, surrounded by lead, while Schuster obtained a value about 12 for the air in the laboratory of Owens College, Manchester.
In order to measure the amount of the excited activity from the tank, a central insulated wire was charged negatively to about 10,000 volts by a Wimshurst machine. After two hours, the wire was removed and wound on an insulated frame connected with a gold-leaf electroscope. The rate of decay of the activity on the wire was found to be about the same as for the excited activity produced by the radium emanation. In order to estimate the amount of 525radium emanation present in the large tank, special experiments were made with a smaller tank in which a known quantity of the radium emanation was introduced by employing a solution of pure radium bromide of known concentration. A central wire was made the negative electrode as before, and, after removal, it was wound on the frame and its activity tested. In this way it was found that the amount of radium emanation present in the large tank, in order to produce the excited activity observed, must have been equal to the equilibrium or maximum amount to be obtained from 9·5 × 10-9 grams of pure radium bromide. The volume of the large tank was 17 cubic metres, so that the amount of emanation present per cubic metre was equivalent to that liberated from 5·6 × 10-10 grams of radium bromide in radio-active equilibrium.
If the amount of the emanation in the tank is taken as the average amount existing in the outside air, the amount of radium emanation present per cubic kilometre of the air is equivalent to that supplied by 0·56 grams of radium bromide.
For the purpose of calculation, suppose the emanation is uniformly distributed over the land portion of the earth (¼ of the total surface), and to extend to an average height of 5 kilometres. The air over the sea is not taken into account as its radio-activity has not been examined. The total amount of emanation present in the atmosphere under these conditions corresponds to that supplied by about 400 tons of radium bromide. In order to maintain this amount of emanation in the atmosphere, it must be supplied at a constant rate from the earth’s surface. Since the greater amount of the emanation probably escapes into the air by transpiration and diffusion through the soil, the emanation cannot reach the surface except from a very thin layer of the earth. The probable thickness of this layer can be estimated if it is assumed that the present loss of heat from the earth is supplied from the radio-active matter contained in it. We have seen (section 271) that, on this hypothesis, there must be an amount of active matter in the earth corresponding to about 300 million tons of radium. If this is supposed to be uniformly distributed, a thickness of layer of about 13 metres will suffice to maintain the calculated amount of emanation in the atmosphere. This thickness of layer is about the order of magnitude to be expected from general considerations.
526These results lead indirectly to the conclusion that a large amount of emanation does undoubtedly exist in the surface crust of the earth.
Experiments were also made by Eve with a large zinc cylinder exposed in the open air. Volume for volume, the average amount of excited activity derived from it was only about one-third of that obtained from the large iron tank. This would reduce the amount of emanation, previously deduced, to about one-third.
Before such calculations can be considered at all definite, it will be necessary to make comparative measurements of the amount of emanation in the atmosphere at various parts of the earth. The air at Montreal is not abnormally active, so that the calculations probably give the right order of magnitude of the quantities.
Eve also observed that the amount of activity to be obtained per unit length of the wire in the zinc cylinder of about 70 cms. in diameter was about the same as for a wire ·5 mms. in diameter charged to 10,000 volts in the open air, supported 20 feet from the ground. This shows that such a potential does not draw in the carriers of excited activity which are more than half a metre away, and probably the range is even less.
It is of great importance to find how large a proportion of the number of ions produced in the atmosphere is due to the radio-active matter distributed throughout it. The results of Eve with the large iron tank, already referred to, indicate that a large proportion of the ionization in the tank was due to the radio-active matter contained in it, for the ratio of the excited activity on the central electrode to the total ionization current in the tank was about ⁷⁄₁₀ of the corresponding ratio for a smaller tank into which a supply of the radium emanation had been introduced.
This result requires confirmation by experiments at other parts of the earth, but the results point to the conclusion that a large part, if not all, of the ionization at the earth’s surface is due to radio-active matter distributed in the atmosphere. A constant rate of production of 30 ions per second per c.c. of air, which has been observed in the open air at the surface of the earth in various localities, would be produced by the presence in each c.c. of the air of the amount of emanation liberated from 2·4 × 10-15 grams of radium bromide in radio-active equilibrium. It is not likely, 527however, that the ionization of the upper part of the atmosphere is due to this cause alone. In order to explain the maintenance of the large positive charge, which generally exists in the upper atmosphere, there must be a strong ionization of the upper air, which may possibly be due to ionizing radiations emitted by the sun.
282. Ionization of atmospheric air. A large number of measurements have been made during the last few years to determine the relative amount of ionization in the atmosphere in different localities and at different altitudes. Measurements of this character were first undertaken by Elster and Geitel with a special type of electroscope. A charged body exposed to the air was attached to a portable electroscope, and the rate of loss of charge was observed by the movement of the gold or aluminium leaf. The rates of discharge of the electroscope for positive and negative electricity were generally different, the ratio depending on the locality and the altitude, and on the meteorological conditions. This apparatus is not suitable for quantitative measurements and the deductions to be drawn from the observations are of necessity somewhat indefinite.
Ebert[430] has designed a portable apparatus in which the number of ions per c.c. of the air can be determined easily. A constant current of air is drawn between two concentric cylinders by means of a fan actuated by a falling weight. The inner cylinder is insulated and connected with an electroscope. Knowing the capacity of the apparatus, and the velocity of the current of air, the rate of movement of the gold-leaf affords a measure of the number of ions present in unit volume of the air drawn between the cylinders.
In this way Ebert found that the number of ions in the air was somewhat variable, but on an average corresponded to about 2600 per c.c. in the particular locality where the measurements were made.
This is the equilibrium number of ions present per c.c. when the rate of production balances the rate of recombination. If q is the number of ions produced per second per unit volume of the air 528and n is the equilibrium number, then q = αn2 where α is the constant of recombination (section 30).
By a slight addition to the apparatus of Ebert, Schuster[431] has shown that the constant of recombination for the particular sample of air under investigation can be determined. The value so obtained for air in the neighbourhood of Manchester was variable, and two or three times as great as for dust-free air. The results of some preliminary measurements showed that the number of ions present per c.c. of the air in different localities varied from 2370 to 3660, while the value of q, the number of ions produced per c.c. per second, varied between 12 and 38·5.
Rutherford and Allan and Eberts showed that the ions in the air had about the same mobility as the ions produced in air by Röntgen rays and radio-active substances. In some recent determinations by Mache and Von Schweidler[432], the velocity of the positive ion was found to be about 1·02 cms. per second, and that of the negative 1·25 cms., for a potential gradient of one volt per cm.
Langevin[433] has recently shown that in addition to these swift moving ions, there are also present in the atmosphere some ions which travel extremely slowly in an electric field. The number of these slowly moving ions in the air in Paris is about 40 times as great as the number of the swifter ions. This result is of great importance, for in the apparatus of Ebert these ions escape detection, since the electric field is not strong enough to carry them to the electrodes during the time of their passage between the cylinders.
283. Radio-activity of ordinary materials. It has been shown that radio-active matter seems to be distributed fairly uniformly over the surface of the earth and in the atmosphere. The very important question arises whether the small radio-activity observed is due to known or unknown radio-elements present in the earth and atmosphere, or to a feeble radio-activity of matter in general, which is only readily detectable when large quantities of matter are present. The experimental evidence is not yet 529sufficient to answer this question, but undoubted proof has been obtained that many of the metals show a very feeble radio-activity. Whether this radio-activity is due to the presence of a slight trace of the radio-elements or is an actual property of the metals themselves will be discussed in more detail in section 286.
Schuster[434] has pointed out that every physical property hitherto discovered for one element has been found to be shared by all the others in varying degrees. For example, the property of magnetism is most strongly marked in iron, nickel, and cobalt, but all other substances are found to be either feebly magnetic or diamagnetic. It might thus be expected on general principles that all matter should exhibit the property of radio-activity in varying degrees. On the view developed in chapter X., the presence of this property is an indication that the matter is undergoing change accompanied by the expulsion of charged particles. It does not, however, by any means follow that because the atom of one element in the course of time becomes unstable and breaks up, that, therefore, the atoms of all the other elements pass through similar phases of instability.
It has already been mentioned (section 8), that Mme Curie made a very extensive examination of most of the elements and their compounds for radio-activity. The electric method was used, and any substance possessing an activity of ¹⁄₁₀₀ of that of uranium would certainly have been detected. With the exception of the known radio-elements and the minerals containing uranium and thorium, no other substances were found to be radio-active even to that degree.
Certain substances like phosphorus[435] possess the property of ionizing a gas under special conditions. The air which is drawn over the phosphorus is conducting, but it has not yet been settled whether this conductivity is due merely to ions formed at the surface of the phosphorus or to ions produced by the phosphorus nuclei or emanations, as they have been termed, which are carried along with the current of air. It does not however appear that the ionization of the gas is in any way due to the presence of a penetrating type of radiation such as is emitted by the radio-active 530bodies. Le Bon (section 8) observed that quinine sulphate, after being heated to a temperature below the melting point and then allowed to cool, showed for a time strong phosphorescence and was able rapidly to discharge an electroscope. The discharging action of quinine sulphate under varying conditions has been very carefully examined by Miss Gates[436]. The ionization could not be observed through thin aluminium foil or gold-leaf, but appeared to be confined to the surface of the sulphate. The current observed by an electrometer was found to vary with the direction of the electric field, indicating that the positive and negative ions had very different mobilities. The discharging action appears to be due either to an ionization of the gas very close to the surface by some short ultra-violet light waves, accompanying the phosphorescence, or to a chemical action taking place at the surface.
Thus, neither phosphorus nor quinine sulphate can be considered to be radio-active, even under the special conditions when they are able to discharge an electrified body. No evidence in either case has been found that the ionization is due to the emission of a penetrating radiation.
No certain evidence has yet been obtained that any body can be made radio-active by exposure to Röntgen rays or cathode rays. A metal exposed to the action of Röntgen rays gives rise to a secondary radiation which is very readily absorbed in a few centimetres of air. It is possible that this secondary radiation may prove to be analogous in some respects to the α rays from the radio-elements. The secondary radiation, however, ceases immediately the Röntgen rays are cut off. Villard[437] stated that a piece of bismuth produced a feeble photographic action after it had been exposed for some time to the action of the cathode rays in a vacuum. It has not however been shown that the bismuth gives out rays of a character similar to those of the radio-active bodies. The experiments of Ramsay and Cooke on the production of apparent activity in inactive matter by the radiations from radium have already been discussed in section 264.
The existence of a very feeble radio-activity of ordinary matter has been deduced from the study of the conductivity of gases in 531closed vessels. The conductivity is extremely minute, and special methods are required to determine it with accuracy. A brief account will now be given of the gradual growth of our knowledge on this important question.
284. Conductivity of air in closed vessels. Since the time of Coulomb onwards several investigators have believed that a charged conductor placed inside a closed vessel lost its charge more rapidly than could be explained by the conduction leak across the insulating support. Matteucci, as early as 1850, observed that the rate of loss of charge was independent of the potential. Boys, by using quartz insulators of different lengths and diameters, arrived at the conclusion that the leakage must in part take place through the air. This loss of charge in a closed vessel was believed to be due in some way to the presence of dust particles in the air.
On the discovery that gases become temporary conductors of electricity under the influence of Röntgen rays and the rays from radio-active substances, attention was again drawn to this question. Geitel[438] and C. T. R. Wilson[439] independently attacked the problem, and both came to the conclusion that the loss of charge was due to a constant ionization of the air in the closed vessel. Geitel employed in his experiments an apparatus similar to that shown in Fig. 103. The loss of charge of an Exner electroscope, with the cylinder of wire netting Z attached, was observed in a closed vessel containing about 30 litres of air. The electroscope system was found to diminish in potential at the rate of about 40 volts per hour, and this leakage was shown not to be due to a want of insulation of the supports.
Wilson, on the other hand, used a vessel of very small volume, in order to work with air which could be completely freed from dust. In the first experiments a silvered glass vessel with a volume of only 163 c.c. was employed. The experimental arrangement is shown in Fig. 104.
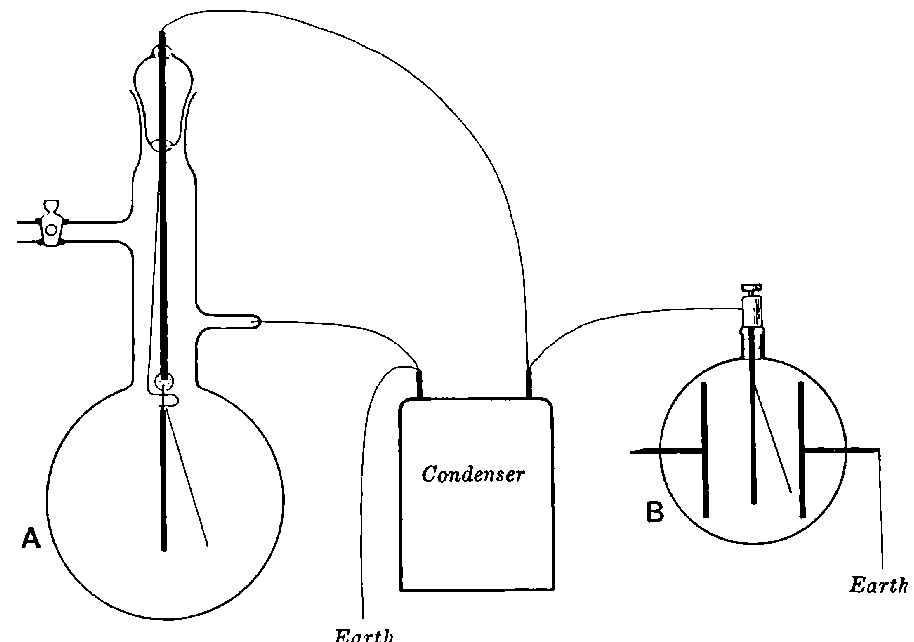
Fig. 104.
The conductor, of which the loss of charge was to be measured, was placed near the centre of the vessel A. It consisted of a 532narrow strip of metal with a gold-leaf attached. The strip of metal was fixed to the upper rod by means of a small sulphur bead. The upper rod was connected with a sulphur condenser with an Exner electroscope B attached to indicate its potential. The gold-leaf system was initially charged to the same potential as the upper rod and condenser by means of a fine steel wire which was caused to touch the gold-leaf system by the attraction of a magnet brought near it. The rate of movement of the gold-leaf was measured by means of a microscope provided with a micrometer eye-piece. By keeping the upper rod at a slightly higher potential than the gold-leaf system, it was ensured that the loss of charge of the gold-leaf system should not be due in any way to a conduction leakage across the sulphur bead.
The method employed by Wilson in these experiments is very certain and convenient when an extremely small rate of discharge is to be observed. In this respect the electroscope measures with certainty a rate of loss of charge much smaller than can be measured by a sensitive electrometer.
533Both Geitel and Wilson found that the leakage of the insulated system in dust-free air was the same for a positive as for a negative charge, and was independent of the potential over a considerable range. The leakage was the same in the dark as in diffuse daylight. The independence of leakage of the potential is strong evidence that the loss of charge is due to a constant ionization of the air. When the electric field acting on the gas exceeds a certain value, all the ions are carried to the electrodes before recombination occurs. A saturation current is reached, and it will be independent of further increase of the electric field, provided, of course, a potential sufficiently high to cause a spark to pass is not applied.
C. T. R. Wilson has recently devised a striking experiment to show the presence of ions in dust-free air which is not exposed to any external ionizing agency. Two large metal plates are placed in a glass vessel connected with an expansion apparatus similar to that described in section 34. On expanding the air, the presence of the ions is shown by the appearance of a slight cloud between the plates. These condensation nuclei carry an electric charge, and are apparently similar in all respects to the ions produced in gases by X rays, or by the rays from active substances.
Wilson found that the loss of charge of the insulated system was independent of the locality. The rate of discharge was unaltered when the apparatus was placed in a deep tunnel, so that it did not appear that the loss of charge was due to an external radiation. From experiments already described, however (section 279), it is probable that about 30 per cent. of the rate of discharge observed was due to a very penetrating radiation. This experiment of Wilson’s indicates that the intensity of the penetrating radiation was the same in the tunnel as at the earth’s surface. Wilson found that the ionization of the air was about the same in a brass vessel as in one of glass, and came to the conclusion that the air was spontaneously ionized.
Using a brass vessel of volume about 471 c.c., Wilson determined the number of ions that must be produced in air per unit volume per second, in order to account for the loss of charge of the insulated system. The leakage system was found to have a capacity of about 1·1 electrostatic units, and lost its 534charge at the rate of 4·1 volts per hour for a potential of 210 volts, and 4·0 volts per hour for a potential of 120 volts. Taking the charge on an ion as 3·4 × 10-10 electrostatic units, this corresponds to a production of 26 ions per second.
Rutherford and Allan[440] repeated the results of Geitel and Wilson, using an electrometer method. The saturation current was observed between two concentric zinc cylinders of diameter 25·5 and 7·5 cms. respectively and length 154 cms. It was found that the saturation current could practically be obtained with a potential of a few volts. Saturation was however obtained with a lower voltage after the air had remained undisturbed in the cylinders for several days. This was probably due to the gradual settling of the dust originally present in the air.
Later observations of the number of ions produced in air in sealed vessels have been made by Patterson[441], Harms[442], and Cooke[443]. The results obtained by different observers are shown in the following table. The value of the charge on an ion is taken as 3·4 × 10-10 electrostatic units:
| Material of vessel | Number of ions produced per c.c. per second | Observer |
|---|---|---|
| Silvered glass | 36 | C. T. R. Wilson |
| Brass | 26 | „ „ |
| Zinc | 27 | Rutherford and Allan |
| Glass | 53 to 63 | Harms |
| Iron | 61 | Patterson |
| Cleaned brass | 10 | Cooke |
It will be shown later that the differences in these results are probably due to differences in the radio-activity of the containing vessel.
285. Effect of pressure and nature of gas. C. T. R. Wilson (loc. cit.) found that the rate of leakage of a charged conductor 535varied approximately as the pressure of the air between the pressures examined, viz. 43 mms. and 743 mms. of mercury. These results point to the conclusion that, in a good vacuum, a charged body would lose its charge extremely slowly. This is in agreement with an observation of Crookes, who found that a pair of gold-leaves retained their charge for several months in a high vacuum.
Wilson[444] at a later date investigated the leakage for different gases. The results are included in the following table, where the ionization produced in air is taken as unity:
| Gas | Relative ionization | (Relative ionization) / (density) |
|---|---|---|
| Air | 1·00 | 1·00 |
| Hydrogen | 0·184 | 2·7 |
| Carbon dioxide | 1·69 | 1·10 |
| Sulphur dioxide | 2·64 | 1·21 |
| Chloroform | 4·7 | 1·09 |
With the exception of hydrogen, the ionization produced in different gases is approximately proportional to their density. The relative ionization is very similar to that observed by Strutt (section 45) for gases exposed to the influence of the α and β rays from radio-active substances, and points to the conclusion that the ionization observed may be due either to a radiation from the walls of the vessel or from external sources.
Jaffé[445] has made a careful examination of the natural ionization in the very heavy gas nickel-carbonyl, Ni(CO)4, in a small silvered glass vessel. The ionization of this gas was 5·1 times that of air at normal pressure while its density is 5·9 times that of air. The leak of the electroscope was nearly proportional to the pressures except at low pressure, when the leak was somewhat greater than would be expected if the pressure law held. The fact that a gas of such high density and complicated structure behaves like the simpler and lighter gases is a strong indication that the ionization itself is due to a radiation from the walls of the vessel and not to a spontaneous ionization of the gas.
536Patterson[446] examined the variation of the ionization of air with pressure in a large iron vessel of diameter 30 cms. and length 20 cms. The current between a central electrode and the cylinder was measured by means of a sensitive Dolezalek electrometer. He found that the saturation current was practically independent of the pressure for pressures greater than 300 mms. of mercury. Below a pressure of 80 mms. the current varied directly as the pressure. For air at atmospheric pressure, the current was independent of the temperature up to 450° C. With further increase of temperature, the current began to increase, and the increase was more rapid when the central electrode was charged negatively than when it was charged positively. This difference was ascribed to the production of positive ions at the surface of the iron vessel. The results obtained by Patterson render it very improbable that the ionization observed in air is due to a spontaneous ionization of the enclosed air: for we should expect the amount of this ionization to depend on the temperature of the gas. On the other hand, these results are to be expected if the ionization of the enclosed air is mainly due to an easily absorbed radiation from the walls of the vessel. If this radiation had a penetrating power about equal to that observed for the α rays of the radio-elements, the radiation would be absorbed in a few centimetres of air. With diminution of pressure, the radiations would traverse a greater distance of air before complete absorption, but the total ionization produced by the rays would still remain about the same, until the pressure was reduced sufficiently to allow the radiation to traverse the air space in the vessel without complete absorption. With still further diminution of pressure, the total ionization produced by the radiation, and in consequence the current observed, would vary directly as the pressure.
286. Examination of ordinary matter for radio-activity. Strutt[447], McLennan and Burton[448], and Cooke[449], independently observed 537about the same time that ordinary matter is radio-active to a slight degree. Strutt, by means of an electroscope, observed that the ionization produced in a closed vessel varied with the material of the vessel. A glass vessel with a removable base was employed and the vessel was lined with the material to be examined. The following table shows the relative results obtained. The amount of leakage observed is expressed in terms of the number of scale divisions of the eye-piece passed over per hour by the gold-leaf:
| Material of lining of vessel | Leakage in scale divisions per hour |
|---|---|
| Tinfoil | 3·3 |
| „ another sample | 2·3 |
| Glass coated with phosphoric acid | 1·3 |
| Silver chemically deposited on glass | 1·6 |
| Zinc | 1·2 |
| Lead | 2·2 |
| Copper (clean) | 2·3 |
| „ (oxidized) | 1·7 |
| Platinum (various samples) | 2·0, 2·9, 3·9 |
| Aluminium | 1·4 |
There are thus marked differences in the leakage observed for different materials and also considerable differences in different samples of the same metal. For example, one specimen of platinum caused nearly twice the leakage of another sample from a different stock.
McLennan and Burton, on the other hand, measured by means of a sensitive electrometer the ionization current produced in the air in a closed iron cylinder 25 cms. in diameter and 130 cms. in length, in which an insulated central electrode was placed. The open cylinder was first exposed for some time at the open window of the laboratory. It was then removed, the top and bottom closed, and the saturation current through the gas determined as soon as possible. In all cases it was observed that the current diminished for two or three hours to a minimum and then very slowly increased again. In one experiment, for example, the initial current observed corresponded to 30 on an arbitrary scale. In the course of four hours the current fell to a minimum of 6·6, and 53844 hours later had risen to a practical maximum of 24. The initial decrease observed is probably due to a radio-activity of the enclosed air or walls of the vessel, which decayed rapidly with the time. The decay of the excited activity produced on the interior surface of the cylinder when exposed to the air was probably responsible for a part of the decrease observed. McLennan ascribes the increase of current with time to a radio-active emanation which is given off from the cylinder, and ionizes the enclosed air. On placing linings of lead, tin, and zinc in the iron cylinder, considerable differences were observed both for the minimum current and also for the final maximum. Lead gave about twice the current due to zinc, while tin gave an intermediate value. These results are similar in character to those obtained by Strutt.
McLennan and Burton also investigated the effect of diminution of pressure on the current. The cylinder was filled with air to a pressure of 7 atmospheres, and allowed to stand until the current reached a constant value. The air was then allowed to escape and the pressure reduced to 44 mms. of mercury. The current was found to vary approximately as the pressure over the whole range. These results are not in agreement with the results of Patterson already described, nor with some later experiments of Strutt. McLennan’s results however point to the conclusion that the ionization was mainly due to an emanation emitted from the metal. Since the air was rapidly removed, a proportionate amount of the emanation would be removed also, and it might thus be expected that the current would vary directly as the pressure. If this is the case the current through the gas at low pressures should increase again to a maximum if time is allowed for a fresh emanation to form.
H. L. Cooke, using an electroscopic method, obtained results very similar to those given by Strutt. Cooke observed that a penetrating radiation was given out from brick. When a brass vessel containing the gold-leaf system was surrounded by brick, the discharge of the electroscope was increased by 40 to 50 per cent. This radiation was of about the same penetrating power as the rays from radio-active substances. The rays were completely absorbed by surrounding the electroscope with a sheet of lead 5392 mms. in thickness. This result is in agreement with the observation of Elster and Geitel, already mentioned, that radio-active matter was present in clay freshly dug up from the earth.
Cooke also observed that the ionization of the air in a brass electroscope could be reduced to about one-third of its usual value if the interior surface of the brass was carefully cleaned. By removing the surface of the brass he was able to reduce the ionization of the enclosed air from 30 to 10 ions per c.c. per second. This is an important observation, and indicates that a large proportion of the radio-activity observed in ordinary matter is due to a deposit of radio-active matter on its surface. It has already been shown that bodies which have been exposed in the presence of the radium emanation retain a residual activity which decays extremely slowly. There can be no doubt that the radium emanation is present in the atmosphere, and the exposed surface of matter, in consequence, will become coated with an invisible film of radio-active matter, deposited from the atmosphere. On account of the slow decay of this activity it is probable that the activity of matter exposed in the open air would steadily increase for a long interval. Metals, even if they are originally inactive, would thus acquire a fairly permanent activity, but it should be possible to get rid of this by removing the surface of the metal or by chemical treatment. The rapid increase of activity of all matter left in a laboratory in which a large quantity of emanation has been released has been drawn attention to by Eve[450]. This superficial activity, due to the products radium D, E, and F, was mainly removed by placing the metal in strong acid.
A number of experiments have been made by J. J. Thomson, N. R. Campbell, and A. Wood in the Cavendish laboratory to examine whether the radio-activity observed in ordinary matter is a specific property of such matter or is due to the presence of some radio-active impurity. An account of these experiments was given by Professor J. J. Thomson in a discussion on the Radio-activity of Ordinary Matter at the British Association meeting at Cambridge, 1904. The results[451], as a whole, support the view that each substance gives out a characteristic type or types of radiation and 540that the radiation is a specific property of the substance. J. J. Thomson[452] has made experiments to observe the action of different substances in cutting off the external very penetrating radiation (section 279) observed by Cooke and McLennan. He found that some substances cut off this external radiation, while others had little if any effect. For example, the ionization in a closed vessel was reduced 17 per cent. by surrounding it with a thick lead envelope; but, on surrounding it with an equivalent absorbing thickness of water, or water mixed with sand, no sensible diminution was observed. In other experiments Wood[453] found that the diminution of the ionization by a given screen depended upon the material of the vessel. For example, the ionization in a lead vessel, surrounded by a lead screen, was reduced 10 per cent., while in an iron vessel it was reduced 24 per cent. He concludes from his experiments that the ionization observed in a closed vessel has a threefold origin. Part of it is due to an external penetrating radiation, part to a secondary radiation set up by it, while the remainder is due to an intrinsic radiation from the walls, altogether independent of the external radiation.
In some experiments of Campbell[454], the variation of the ionization current between two parallel plates was observed for a progressive increase of the distance between them. The effects observed are shown in Fig. 105. The curves at first rise rapidly, then bend over and finally become a straight line. The knee of the curve is at a different distance for the different substances. The shape of these curves indicates that two types of radiation are present, one of which is readily absorbed in the gas while the other, a more penetrating type of radiation, extends over the whole distance between the plates. In another series of experiments, one side of the testing vessel was of thin aluminium, and the ionization current was observed when an exterior screen was brought up to it. Lead gave a considerable increase, but the radiation from it was readily absorbed by an interposed screen. The radiation emitted by carbon and zinc was more than twice as penetrating as from lead.
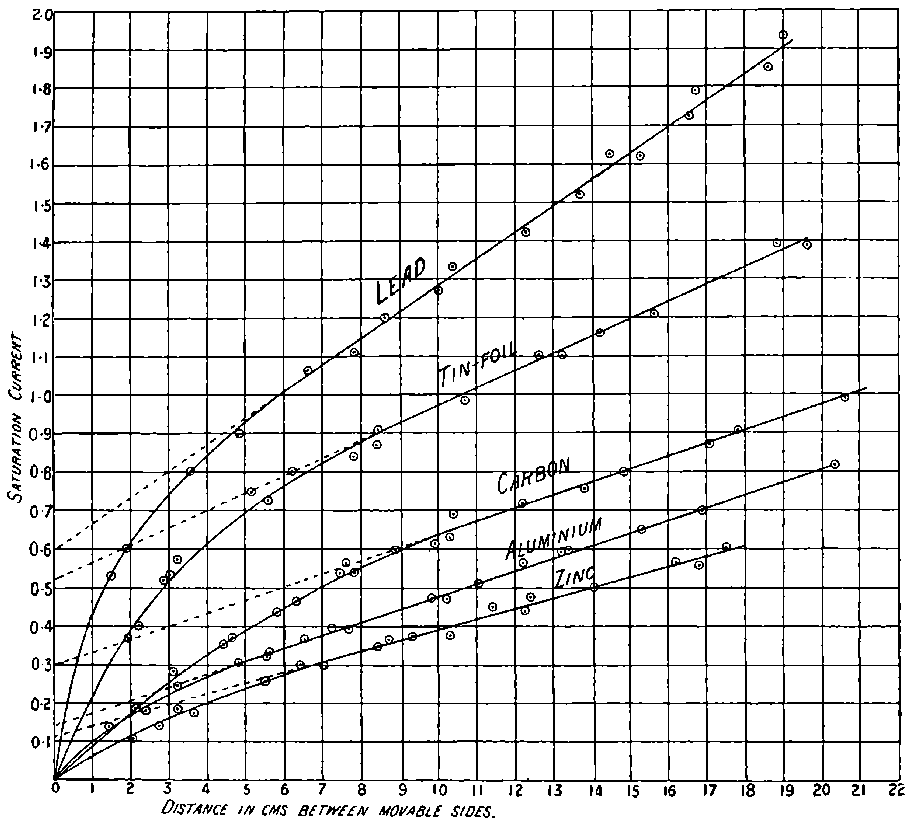
Fig. 105.
Attempts were made to see whether a radio-active emanation was given off by dissolving solid substances and then keeping the solutions in a closed vessel and afterwards testing the activity of the air drawn from them. In some cases an emanation was observed, but the amount varied with different specimens of the same material; in others no effect was detected.
When linings of different substances were placed in a closed testing vessel, the ionization current in most cases fell at first, passed through a minimum, and then slowly increased to a maximum. For lead the maximum was reached in 9 hours, for tin in 14 and for zinc in 18 hours. These results indicate that an emanation is given off from the metal, and that the amount reaches a maximum value at different intervals in the various cases. This was confirmed by an examination of a piece of lead which was left 542in radium-free nitric acid. Twenty times the normal effect was observed after this treatment. This is probably due to the increase of porosity of the lead which allows a greater fraction of the emanation produced in the metal to diffuse out with the gas.
The activity observed in ordinary matter is extremely small. The lowest rate of production of ions yet observed is 10 per cubic centimetre per second in a brass vessel. Suppose a spherical brass vessel is taken of capacity 1 litre. The area of the interior surface would be about 480 sq. cms. and the total number of ions produced per second would be about 104. Now it has been shown, in section 252, that an α particle projected from radium itself gives rise to 8·6 × 104 ions before it is absorbed in the gas. An expulsion of one α particle every 8 seconds from the whole vessel, or of one α particle from each square centimetre of surface per hour would thus account for the minute conductivity observed. Even if it were supposed that this activity is the result of a breaking up of the matter composing the vessel, the disintegration of one atom per second per gram, provided it was accompanied by the expulsion of an α particle, would fully account for the conductivity observed.
While the experiments, already referred to, afford strong evidence that ordinary matter does possess the property of radio-activity to a feeble degree, it must not be forgotten that the activity observed is excessively minute, compared even with a weak radio-active substance like uranium or thorium. The interpretation of the results is complicated, too, by the presence of the radium emanation in the atmosphere, for we have seen that the surface of every body exposed to the open air must become coated with the slowly changing transformation products of the radium emanation. The distribution of radio-active matter throughout the constituents of the earth renders it difficult to be certain that any substance, however carefully prepared, is freed from radio-active impurities. If matter in general is radio-active, it must be undergoing transformation at an excessively slow rate, unless it be supposed (see Appendix A) that changes of a similar character to those observed in the radio-elements may occur without the appearance of their characteristic radiations.
A brief account is given here of some investigations made by the writer on the properties of the α rays from radium—investigations which were not completed in time for the results to be incorporated in the text.
The experiments were undertaken primarily with a view of determining accurately the value of e/m of the α particle from radium, in order to settle definitely whether or not it is an atom of helium. In the previous experiments of the writer, Becquerel, and Des Coudres, on this subject (sections 89, 90, and 91), a thick layer of radium in radio-active equilibrium has been used as a source of α rays. Bragg (section 103) has shown that the rays emitted from radium under such conditions are complex, and consist of particles projected over a considerable range of velocity. In order to obtain a homogeneous pencil of rays it is necessary to use a very thin layer of a simple radio-active substance as a source of rays. In the experiments that follow, this condition was fulfilled by using a fine wire which was made active by exposure for several hours in the presence of a large quantity of radium emanation. By charging the wire negatively the active deposit was concentrated upon the wire, which was made intensely active. The active deposit initially contains radium A, B, and C. The activity of radium A practically disappears in about fifteen minutes, and the α radiation is then due entirely to the single product radium C, since radium B is a rayless product. The activity of radium C decreases to about 15 per cent. of its initial value after two hours.
Magnetic deflection of the α rays. The photographic method was employed to determine the deviation of the pencil of rays in a magnetic field. The experimental arrangement is shown in Fig. 106. The rays from the active wire, which was placed in a slot, passed through a narrow slit and fell normally on a photographic plate, placed at a known distance above the slit. The apparatus was enclosed in a 544brass tube which could be exhausted rapidly to a low pressure by means of a Fleuss pump. The apparatus was placed in a strong uniform magnetic field parallel to the plane of the slit. The magnetic field was reversed every ten minutes, so that on developing the plate two narrow bands were observed, the distance between which represented twice the deviation from the normal of the pencil of rays by the magnetic field. The width of the band was found to be the same whether the magnetic field was applied or not, showing that the pencil of rays was homogeneous and consisted of α particles projected with the same velocity.
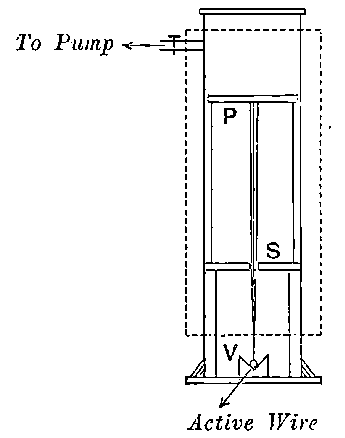
Fig. 106.
By placing the photographic plate at different distances from the slit it was found that the rays, after entering the magnetic field, described the arc of a circle of radius ρ equal to 42·0 cms. The strength of field H was 9470 C.G.S. units, so that the value of Hρ for the α particles expelled from radium C is 398,000. This is in good agreement with the maximum values of Hρ, previously found for radium rays (see section 92).
The electric deviation of the rays from radium C has not yet been accurately measured, but an approximate determination of e/m for the α particles can be obtained by assuming that the heating effect of radium C is a measure of the kinetic energy of the α particles expelled from it. We have seen in section 246 that the heating effect of the radium C present in one gram of radium in radio-active equilibrium is 31 gram calories per hour, which corresponds to an emission of energy of 3·6 × 105 ergs per second. Now when radio-active equilibrium is reached, the number of α particles expelled from radium C per second is equal to the number of α particles expelled per second from radium at its minimum activity. This number, n, is 6·2 × 1010 (section 93).
substituting the value of n, and the value of the ionic charge e. The value of e in this case has not been assumed, since n = i/e, where 545i was the measured current due to the charge carried by the α rays.
From the magnetic deflection, it is known that
From these two equations we obtain
These values are in surprisingly good agreement with the previous values of the writer and Des Coudres (section 91). On account of the uncertainty attaching to the value of n, not much weight can be attached to the determination by this method of the constants of the α particles.
Decrease of velocity of the α particles in passing through matter. Some experiments were made to determine the velocity of the α particles from radium C after passing through known thicknesses of aluminium. The previous apparatus was employed, and the distance between the photographic bands was observed for successive layers of aluminium foil, each ·00031 cms. thick, placed over the active wire. The photographic plate was placed 2 cms. above the slit, and the magnetic field extended 1 cm. below the slit. The amount of deviation of the rays is inversely proportional to their velocity after traversing the aluminium screens. The impressions on the plate were clear and distinct, and about the same in all cases, showing that the rays were still homogeneous after passing through the aluminium.
A clear photographic impression was obtained for 12 layers of foil, but it was not found possible to obtain any effect through 13 layers. This result shows that the photographic action of the rays, like the ionizing action, ceases very abruptly.
The results obtained are shown in the following table. Assuming that the value of e/m is constant, the third column gives the velocity of the α particles after traversing the aluminium. This is expressed in terms of V₀, the velocity of the α particle when the screens are removed.
| Number of layers of aluminum foil | Distance between bands on the plate | Velocity of α particles |
|---|---|---|
| 0 | 1·46 mms. | 1·00 V₀ |
| 5 | 1·71 „ | ·85 „ |
| 8 | 1·91 „ | ·76 „ |
| 10 | 2·01 „ | ·73 „ |
| 12 | 2·29 „ | ·64 „ |
| 13 | No photographic effect |
546The velocity of the α particle is thus reduced only about 36 per cent. of its initial value when it fails to produce any action on the photographic plate.
Now Bragg has shown (section 104) that the α particle produces approximately the same number of ions per cm. of path in air over its whole range. Consequently, the simplest assumption to make is that the energy of the α particle is diminished by a constant amount in traversing each layer of foil. After passing through 12 layers the kinetic energy is reduced to 41 per cent. of the maximum. Each layer of foil thus absorbs 4·9 per cent. of the maximum energy. The observed kinetic energy of the α particle after passing through successive layers of foil, and the value calculated on the above assumptions, are shown in the following table.
| Number of layers of aluminum foil | Observed energy | Calculated energy |
| 0 | 100 | 100 |
| 5 | 73 | 75 |
| 8 | 58 | 61 |
| 10 | 53 | 51 |
| 12 | 41 | 41 |
The experimental and theoretical values agree within the limits of experimental error. We may thus conclude, as a first approximation, that the same proportion of the total energy is abstracted from the α particles in passing through equal distances of the absorbing screen.
Range of ionization and photographic action in air. The abrupt falling off of the photographic impression after the rays had passed through 12 layers of foil suggested that it might be directly connected with the corresponding abrupt falling off of the ionization in air, so clearly brought out by Bragg. This was found to be the case. It was found experimentally that the absorption in each layer of aluminium foil was equivalent to that produced by a distance of ·54 cms. of air. Twelve layers of foil thus corresponded to 6·5 cms. of air. Now Bragg found that the α rays from radium C ionize the air for a distance 6·7 cms., and that the ionization then falls off very rapidly. We may thus conclude that the α rays cease to affect the photographic plate at the same velocity as that at which they cease to ionize the gas. This is a very important result, and, as we shall see later, suggests that the action on the photographic plate is due to an ionization of the photographic salts.
547The velocity of the α particles from the different radio-active products can at once be calculated, knowing the maximum range in air of the α rays from each product. The latter have been experimentally determined by Bragg. The velocity is expressed in terms of V₀, the initial velocity of the α particles from radium C. The rays from radium C are projected with a greater velocity than the rays from the other products of radium.
| Product | Maximum range of α particles in air | Velocity of α particles |
|---|---|---|
| Radium | 3 cms. | ·82 V₀ |
| Emanation | 3·8 or 4·4 cms. | ·87 or ·90 V₀ |
| Rad. A | 4·4 or 3·8 „ | ·90 or ·87 V₀ |
| Rad. C | 6·7 „ | 1·00 V₀ |
It is difficult to determine from the experiments whether the range 3·8 cms. belongs to the rays from the emanation or from radium A. The mean velocity of the α particles is thus ·90 V₀, and the maximum variation for the individual products does not vary more than 10 per cent. from the mean value.
The results of Becquerel, discussed in section 92, at once receive an explanation on the above results. The α particles, expelled from radium in radio-active equilibrium, have all ranges lying between 0 and 6·7 cms. of air. The velocity of the α particles which are able to produce a photographic impression varies between ·64 V₀ and V₀. The particles which have only a short range in air are projected with a smaller velocity than those which have a greater range. The former are in consequence more bent by a magnetic field. It is thus to be expected that the apparent curvature of the path of rays in a uniform magnetic field will be greater close to the radium than at some distance away.
Range of phosphorescent action in air. Some experiments were also made to see whether the action of the α rays in producing luminosity in substances like zinc sulphide, barium platinocyanide, and willemite, ceased at the same distance as the ionizing action.
A very active wire was placed on a moveable plate, the distance of which from a fixed screen of phosphorescent substance could be varied. The distance at which the phosphorescent action ceased could be determined fairly accurately. Different thicknesses of aluminium foil were then placed over the active wire, and the corresponding distance at which the luminosity disappeared was 548measured. The results are shown graphically in Fig. 107, where the ordinates represent the distance of the phosphorescent screen from the active wire, and the abscissae the number of layers of aluminium foil, each ·00031 cms. thick.
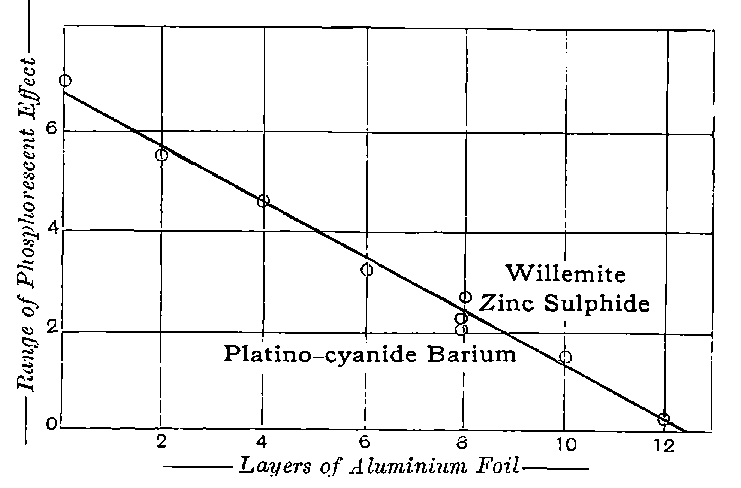
Fig. 107.
It is seen that the curve joining the points is a straight line. 12·5 thicknesses of foil absorbed the rays to the same extent as 6·8 cms. of air, so that each thickness of aluminium corresponded in absorbing power to ·54 cms. of air. For a screen of zinc sulphide, the phosphorescent action ceased at a distance of air of 6·8 cms., showing that the photographic and phosphorescent ranges of the α rays in air were practically identical.
The experiments with barium platinocyanide and willemite were more difficult, as the β and γ rays from the active wire produced a luminosity comparable with that produced by the α rays. Fairly concordant results, however, were obtained by introducing a thin sheet of black paper between the active wire and the screen. If the luminosity was sensibly changed, it was concluded that the α rays still produced an effect, and in this way the point of cessation of phosphorescent action could be approximately determined. For example, with eight thicknesses of foil over the active wire the additional thickness of air required to cut off the phosphorescent effect of the a rays was 2·5 cms. for willemite, and 2·1 cms. for barium platinocyanide.
549The corresponding distance for zinc sulphide was 2·40 cms., a value intermediate between the other two.
Since eight layers of foil are equivalent to 4·3 cms. of air, the ranges in air of phosphorescent action for zinc sulphide, barium platinocyanide, and willemite correspond to 6·7, 6·8, and 6·4 cms. respectively. The differences observed are quite likely to be due to experimental error.
Discussion of results. We have seen that the ionizing, phosphorescent, and photographic actions of the α rays emitted from radium C cease after traversing very nearly the same distance of air. This is a surprising result when it is remembered that the α particle, after passing through this depth of air, still possesses a velocity of at least 60 per cent. of its initial value. Taking the probable value of the initial velocity of the α particle from radium C as 2·5 × 109 cms. per sec., the ionizing, phosphorescent, and photographic actions cease when the velocity of the α particle falls below 1·5 × 109 cms. per second, that is, a velocity of about ¹⁄₂₀ of that of light. The particle still possesses nearly 40 per cent. of its initial energy of projection at this stage.
These results show that the property of the α rays of producing ionization in gases, of producing luminosity in some substances, and of affecting a photographic plate, ceases when the velocity of the α particle falls below a certain fixed value which is the same in each case. It seems reasonable, therefore, to suppose that these three properties of the α rays must be ascribed to a common cause. Now the absorption of the α rays in gases is mainly a consequence of the energy absorbed in the production of ions in the gas. When the α particles are completely absorbed in the gas, the same total amount of ionization is produced, showing that the energy required to produce an ion is the same for all gases. On the other hand, for a constant source of radiation, the ionization per unit volume of the gas is approximately proportional to its density. Since the absorption of the α rays in solid matter is approximately proportional to the density of the absorbing medium compared with air, it is probable that this absorption is also a result of the energy used up in producing ions in the solid matter traversed, and that about the same amount of energy is required to produce an ion in matter whether solid, liquid, or gaseous.
It is probable, therefore, that the production of ions in the phosphorescent material and in the photographic film would cease at about 550the same velocity for which the α particle is unable to ionize the gas. On this view, then, the experimental results receive a simple explanation. The action of the α rays in producing photographic and phosphorescent actions is primarily a result of ionization. This ionization may possibly give rise to secondary actions which influence the effects observed.
This point of view is of interest in connection with the origin of the “scintillations” observed in zinc sulphide and other substances when exposed to the action of the α rays. This effect is ascribed by Becquerel to the cleavage of the crystals under the bombardment of the α particles. These results, however, show that we must look deeper for the explanation of this phenomenon. The effect is primarily due to the production of ions in the phosphorescent material and not to direct bombardment, for we have seen that the α particle produces no scintillations when it still possesses a large amount of kinetic energy. It seems not unlikely that the scintillations produced by the α rays must be ascribed to the recombination of the ions which are produced by the α particle in the crystalline mass. It is difficult to see how this ionization could result in a cleavage of the crystals.
This close connection of the photographic and phosphorescent actions of the α rays with their property of producing ions, raises the question whether photographic and phosphorescent actions in general may not, in the first place, be due to a production of ions in the substance.
Ionization curve for the α rays from radium C. Mr McClung, working in the laboratory of the writer, has recently determined the relative ionization per unit path of the α particles projected from radium C, using the method first employed by Bragg and discussed in section 104. An active wire, exposed for several hours to the emanation from radium, was used as a source of rays. The α particles were homogeneous, since the film of radio-active matter was extremely thin.
The relation between the ionization observed over the cross section of the narrow cone of rays and the distance from the source of rays is shown in Fig. 108.
Fig. 108.
The curve exhibits the same peculiarities as those given by Bragg for a thin film of matter of one kind. The ionization of the α particle per unit path increases slowly for about 4 cms. There is then a more rapid increase just before the α particle ceases to ionize the 551gas, and then a rapid falling off. The ionization does not appear to end so abruptly as is really the case, since there is a correction to be applied for the angle subtended by the cone of rays. The maximum range of the α rays in air was 6·7 cms., a number in agreement with that obtained by Bragg by measurements on the range of the rays from radium.
These results show that the ionization per unit path of the α particle increases at first slowly and then rapidly with decrease of velocity until the rays cease to ionize the gas.
Energy required to produce an ion. From the above results the energy required to produce an ion by collision of the α particle with the gas molecules can readily be deduced. The α particles, emitted from radium itself, are initially projected with a velocity ·88V₀ 552where V₀ is the initial velocity of projection of the α particles from radium C. The α particles cease to ionize the gas at a velocity ·64V₀. From this it can at once be deduced that ·48 of the total energy of the α particle, shot out by radium itself, is absorbed when it ceases to ionize the gas. Assuming that the heating effect of radium at its minimum activity—25 gram calories per hour per gram—is a measure of the kinetic energy of the expelled α particles, it can be calculated that the kinetic energy of each α particle is 4·7 × 10-6 ergs. The amount of energy absorbed when the α particle just ceases to ionize the gas is 2·3 × 10-6 ergs. Assuming that this energy is used up in ionization, and remembering that the α particle from radium itself produces 86000 ions in its path (section 252), the average energy required to produce an ion is 2·7 × 10-11 ergs. This is equivalent to the energy acquired by an ion moving freely between two points differing in potential by 24 volts.
Townsend found that fresh ions were produced by an electron for a corresponding difference of potential of 10 volts. Stark, from other data, obtained a value 45 volts, while Langevin considers that 60 volts is an average value. The value obtained by Rutherford and McClung for ionization by X-rays was 175 volts, and is probably too high.
Rayless changes. We have seen that the α particles from the radio-active substances are projected with an average velocity not more than 30 per cent. greater than the minimum velocity, below which the α particles are unable to produce any ionizing, photographic, or phosphorescent action. Such a conclusion suggests that the property of the radio-active substances of emitting α particles has been detected because the α particles were projected slightly above this minimum velocity. A similar disintegration of matter may be taking place in other substances at a rate much greater than in uranium without producing much electrical effect, provided the α particles are projected below the critical velocity.
The α particle, on an average, produces about 100,000 ions in the gas before it is absorbed, so that the electrical effect observed is about 100,000 times as great as that due to the charge carried by the α particles alone.
It is not unlikely that the numerous rayless products which have been observed may undergo disintegration of a similar character to the products which obviously emit α rays. In the rayless product the 553α particle may be expelled with a velocity less than 1·5 × 109 cms. per second and so fail to produce much electrical effect.
These considerations have an important bearing on the question whether matter in general is radio-active. The property of emitting α particles above the critical velocity may well be a property only of a special class of substances, and need not be exhibited by matter in general. At the same time the results suggest that ordinary matter may be undergoing transformation accompanied by the expulsion of α particles at a rate much greater than that shown by uranium, without producing appreciable electrical or photographic action.
Those natural mineral substances which possess marked radio-active properties have been found to contain either uranium or thorium, one of these elements being always present in sufficient proportion readily to permit its chemical separation and identification by the ordinary analytical methods[455].
A large number of uranium and thorium minerals are known at the present time, but they are for the most part found very sparingly, and some of them have been observed to occur only in a single locality. The chief commercial sources of uranium are uraninite, gummite, and carnotite, while thorium is obtained almost exclusively from monazite.
Rutherford and Soddy (Phil. Mag. 65, 561 (1903)), were the first to call attention to the important fact that the relations between the various radio-active substances and the other elements could best be determined from the study of the natural minerals in which these bodies occur, since these minerals represent mixtures of extreme antiquity, which have remained more or less undisturbed for almost countless ages. In dealing with these matters, however, it is highly important that we bring to our aid the data furnished by geology and mineralogy, from which it is often possible to determine the relative ages of the different substances with at least a rough degree of approximation. Thus, for example, if a certain mineral occurs as a primary constituent of a rock of remote geological period, it can safely be assumed that its age is greater than that of a similar or different mineral occurring in a later formation. It is, moreover, quite evident that those minerals which are obviously produced by the decomposition and alteration of the primary minerals, through the action of percolating water and other agencies acting from the surface downward, 555are of less antiquity than the primary minerals from which they originated. Through the application of these considerations it should, in general, be possible to arrange the various minerals roughly in the order of their probable ages.
The most familiar and widely known uranium mineral is uraninite, commonly called pitchblende, which consists essentially of uranium dioxide (UO2), uranium trioxide (UO3), and lead oxide (PbO), present in varying proportions. The uraninites can be distinguished as primary, namely, those which occur as a primary constituent of pegmatitic dikes and coarse granites, and secondary, when they occur in metalliferous veins associated with the sulphides of silver, lead, copper, nickel, iron, and zinc. The former varieties are quite frequently crystalline in character, contain a larger proportion of the rare earths and helium, and have a higher specific gravity than the latter, which are always massive and botryoidal.
The following are the most prominent localities in which primary uraninites occur:
1. North Carolina, U.S.A. (especially in Mitchell and Yancey counties). The uraninite is found in a coarse pegmatitic dike which is mined for the mica constituent. The associated feldspar of the dike is considerably decomposed through the action of meteoric waters and gases, and the uraninite itself is largely altered into the secondary minerals gummite and uranophane through the same agencies. Among the associated primary minerals are allanite, zircon, columbite, samarskite, fergusonite and monazite, while the secondary minerals include gummite, thorogummite, uranophane, autunite, phosphuranylite, hatchettolite, and cyrtolite. The geological period of this formation is difficult to establish with certainty, but is stated to be perhaps Archean, or possibly to correspond with the close of the Ordovician or with the Permian.
2. Connecticut, U.S.A. The best known localities are Glastonbury, where the uraninite is found in the feldspar quarries, and Branchville, where it occurs in an albitic granite. Both of these localities have furnished fine crystals. The geological period probably corresponds with the close of the Ordovician or Carboniferous eras, and is stated to be certainly Post-Cambrian and Pre-Triassic. Among the associated minerals are (primary) columbite, (secondary) torbernite and autunite.
3. Southern Norway, particularly in the neighbourhood of Moss. Here uraninite occurs in the augite-syenite and pegmatite. The 556varieties found are known as cleveite and bröggerite, and among the primary associated minerals are orthite, fergusonite, monazite, and thorite. The period is stated to be Post-Devonian.
4. Llano County, Texas. The variety of uraninite known as nivenite is found here in a quartzose pegmatite, associated with the primary minerals gadolinite, allanite and fergusonite, and the secondary minerals cyrtolite, yttrialite, gummite, and thorogummite.
Secondary uraninite is found at Johanngeorgenstadt, Marienberg and Schneeberg in Saxony, at Joachimsthal and Pribam in Bohemia, at Cornwall in England, and at Black Hawk, Colorado, and in the Black Hills, South Dakota, in the United States. The exact geological period of most of these secondary occurrences is somewhat uncertain, but they are undoubtedly very much later than the primary occurrences mentioned above.
As a matter of general interest the analysis of a typical primary uraninite (No. 1) and of a typical secondary uraninite (No. 2) is given below[456]:
| No. 1 Glastonbury, Conn. | No. 2 Johanngeorgenstadt, Saxony | |
|---|---|---|
| Sp. Gr. | 9·59 | 6·89 |
| UO3 | 26·48 | 60·05 |
| UO2 | 57·43 | 22·33 |
| ThO2 | 9·79 | ... |
| CeO2 | 0·25 | ... |
| La2O3 | 0·13 | ... |
| Y2O3 | 0·20 | ... |
| PbO | 3·26 | 6·39 |
| CaO | 0·08 | 1·00 |
| He | und. | und. |
| H2O | 0·61 | 3·17 |
| Fe2O3 | 0·40 | 0·21 |
| SiO2 | 0·25 | 0·50 |
| Al2O3 | ... | 0·20 |
| Bi2O3 | ... | 0·75 |
| CuO | ... | 0·17 |
| MnO | ... | 0·09 |
| MgO | ... | 0·17 |
| Na2O | ... | 0·31 |
| P2O5 | ... | 0·06 |
| SO3 | ... | 0·19 |
| As2O3 | ... | 2·34 |
| Insoluble | 0·70 | ... |
The following list comprises the more important radio-active minerals, with their approximate chemical composition and some notes on their occurrence and probable origin.
| 557 | ||
| Name | Composition | Remarks |
|---|---|---|
| Uraninite, Cleveite, Bröggerite, Nivenite, Pitchblende | Oxides of uranium and lead. Usually contains thorium, other rare earths and helium. Uranium 50-80%. Thorium 0–10% | Occurs primary as a constituent of rocks and secondary in veins with metalliferous sulphides |
| Gummite | (Pb, Ca) U3SiO12 . 6H2O? Uranium 50–65% | An alteration product of uraninite. Formed by the action of percolating waters |
| Uranophane, Uranotil | CaO . 2UO3 . 2SiO2 . 6H2O Uranium 44–56% | An alteration product of uraninite through gummite |
| Carnotite | A vanadate of uranium and potassium. Uranium 42–51% | Occurs as a secondary mineral impregnating a porous, sedimentary sandstone. Found in Colorado and Utah |
| Uranosphaerite | Bi2O3 . 2UO3 . 3H2O. Uranium 41% | Alteration product of other uranium minerals |
| Torbernite, Cuprouranite | CuO . 2UO3 . P2O5 . 8H2O. Uranium 44–51% | „ „ |
| Autunite, Calciouranite | CaO . 2UO3 . P2O5 . 8H2O. Uranium 45–51% | „ „ |
| Uranocircite | BaO . 2UO3 . P2O5 . 8H2O. Uranium 46% | „ „ |
| Phosphuranylite | 3UO3 . P2O5 . 6H2O. Uranium 58–64% | „ „ |
| Zunerite | CuO . 2UO3 . As2O5 . 8H2O. Uranium 46% | „ „ |
| Uranospinite | CaO . 2UO3 . As2O5 . 8H2O. Uranium 49% | „ „ |
| Walpurgite | 5Bi2O3 . 3UO3 . As2O5 . 12H2O. Uranium 16% | „ „ |
| Thorogummite | UO3 . 3ThO2 . 3SiO2 . 6H2O? Uranium 41% | A variety of gummite |
| Thorite, Orangite, Uranothorite | ThSiO4. Uranium 1–10%. Thorium oxide 48–71% | A primary constituent of pegmatite dikes |
| Thorianite | Oxide of thorium, uranium, the rare earths and lead. Contains a relatively large proportion of helium. Uranium 9–10%. Thorium oxide 73–77% | Occurs as a primary constituent of a pegmatite dike in Ceylon. Geological age probably Archean |
| 558Samarskite | Niobate and tantalate of rare earths. Uranium 8–10% | Primary constituent of pegmatite dikes |
| Fergusonite | Metaniobate and tantalate of rare earths. Uranium 1–6% | „ „ |
| Euxenite | Niobate and titanate of rare earths. Uranium 3–10% | „ „ |
| Monazite | Phosphate of the rare earths, chiefly cerium. Uranium 0·3–0·4% | „ „ |
The numbers refer to the pages.
CAMBRIDGE: PRINTED BY JOHN CLAY, M.A. AT THE UNIVERSITY PRESS.
Conduction of Electricity through Gases. By J. J. Thomson, D.Sc., LL.D., Ph.D., F.R.S., Fellow of Trinity College and Cavendish Professor of Experimental Physics. Demy 8vo. viii + 568 pp. 16s.
CONTENTS.
I. Electrical Conductivity of Gases in a normal state.
II. Properties of a Gas when in the conducting state.
III. Mathematical Theory of the Conduction of Electricity through a Gas containing Ions.
IV. Effect produced by a Magnetic Field on the Motion of the Ions.
V. Determination of the Ratio of the Charge to the Mass of an Ion.
VI. Determination of the Charge carried by the Negative Ion.
VII. On some Physical Properties of Gaseous Ions.
VIII. Ionisation by Incandescent Solids.
IX. Ionisation in Gases from Flames.
X. Ionisation by Light. Photo-Electric Effects.
XI. Ionisation by Röntgen Rays.
XII. Becquerel Rays.
XIII. Spark Discharge.
XIV. The Electric Arc.
XV. Discharge through Gases at Low Pressures.
XVI. Theory of the Discharge through Vacuum Tubes.
XVII. Cathode Rays.
XVIII. Röntgen Rays.
XIX. Properties of Moving Electrified Bodies.
Supplementary Notes.
Index.
Times.—“It is difficult to think of a single branch of the physical sciences in which these advances are not of fundamental importance. The physicist sees the relations between electricity and matter laid bare in a manner hardly hoped for hitherto.... The workers in the field of Science are to-day reaping an unparalleled harvest, and we may congratulate ourselves that in this field at least we more than hold our own among the nations of the world.”
A Treatise on the Theory of Solution, including the Phenomena of Electrolysis. By William Cecil Dampier Whetham, M.A., F.R.S., Fellow of Trinity College. Demy 8vo. x + 488 pp. 10s. net.
Athenæum.—“The choice and arrangement of the matter included are alike excellent.... Mr Whetham’s book is probably the most complete and satisfactory treatise on the subject in any language and as such is sure to enjoy a wide popularity.”
Nature.—“The treatment throughout is characterised by great clearness, especially in the physical and mathematical portions, so that the volume may be warmly recommended to students of chemistry.”
CONTENTS OF Mr Whetham’s ‘Solution and Electrolysis.’
I. Thermodynamics.
II. The Phase Rule.
III. The Phase Rule. Two Components. Solutions.
IV. Solubility.
V. Osmotic Pressure.
VI. Vapour Pressures and Freezing Points.
VII. Theories of Solution.
VIII. Electrolysis.
IX. Conductivity of Electrolytes.
X. Galvanic Cells.
XI. Contact Electricity and Polarization.
XII. The Theory of Electrolytic Dissociation.
XIII. Diffusion in Solutions.
XIV. Solutions of Colloids.
Additions.
Table of Electro-chemical Properties of Aqueous Solutions.
Electricity and Magnetism: an Elementary Text-book, Theoretical and Practical. By R. T. Glazebrook, M.A., F.R.S., Director of the National Physical Laboratory and Fellow of Trinity College, Cambridge. Crown 8vo. Cloth. 1–440 pp. 7s. 6d.
Athenæum.—“If the nature of the book be taken into consideration, it will be found unusually free from the influence of the examination spirit. The writing is bright and interesting, and will stimulate a desire, we think, for further study.”
Guardian.—“Every schoolmaster and teacher who has under consideration the selection of a text-book for his better students should most certainly look into this book. The information is everywhere absolutely sound and reliable.”
PREFACE. Some words are perhaps necessary to explain the publication of another book dealing with Elementary Electricity. A considerable portion of the present work has been in type for a long time; it was used originally as a part of the practical work in Physics for Medical Students at the Cavendish Laboratory in connexion with my lectures, and was expanded by Mr Wilberforce and Mr Fitzpatrick in one of their Laboratory Note-books of Practical Physics.
When I ceased to deliver the first year course I was asked to print my lectures for the use, primarily, of the Students attending the practical classes; the lectures on Mechanics, Heat and Light have been in type for some years. Other claims on my time have prevented the issue of the present volume until now, when it appears in response to the promise made several years ago.
Meanwhile the subject has changed; but while this is the case the elementary laws and measurements on which the science is based remain unaltered, and I trust the book may be found of service to others besides my successors at the Cavendish Laboratory.
The book is to be used in the same way as its predecessors. The apparatus for most of the Experiments is of a simple character and can be supplied at no great expense in considerable quantities.
Thus the Experiments should all, as far as possible, be carried out by the members of the class, the teacher should base his reasoning on the results actually obtained by his pupils. Ten or twelve years ago this method was far from common; the importance to a School of a Physical Laboratory is now more generally recognized; it is with the hope that the book may be of value to those who are endeavouring to put the method in practice that it is issued now.
583Heat and Light. An Elementary Text-book, Theoretical and Practical, for Colleges and Schools. By R. T. Glazebrook, M.A. Crown 8vo. 5s.
Also in separate volumes:
Mechanics and Hydrostatics. An Elementary Text-book, Theoretical and Practical, for Colleges and Schools. By R. T. Glazebrook, M.A. Crown 8vo. 8s. 6d.
Also in separate volumes:
EXTRACTS FROM PRESS NOTICES.
“Schools and Colleges will certainly benefit by adopting this book for their students.” Nature.
“Mr Glazebrook’s volumes on Heat and Light deal with these subjects from the experimental side and it is difficult to admire sufficiently the ingenuity and simplicity of many of the experiments without losing sight of the skill and judgment with which they are arranged.” Saturday Review.
“The books almost cover the advanced stages of the South Kensington prospectus and their use can certainly be recommended to all who wish to study these subjects with intelligence and thoroughness.” Schoolmaster.
“Mr Glazebrook’s great practical experience has enabled him to treat the experimental aspect of the book with unusual power and it is in this that the great value of the book as compared with most of the ordinary manuals consists.” Educational Review.
“The book is very simply and concisely written, is clear and methodic in arrangement.... We recommend the book to the attention of all students and teachers of this branch of physical science.” Educational News.
“We wish Mr Glazebrook every success on the extension of his practical system to all the Colleges and Schools of the country. It is the only way in which the interest of the student can be awakened and the study of the subject made popular and real.” Technical World.
“It will be especially appreciated by teachers who possess the necessary apparatus for experimental illustrations.” Athenæum.
“Text-books on this subject are generally too simple or too elaborate for a conception of elementary mechanical principles. This book cannot fail to recommend itself therefore for a first course preliminary to the study of physical science. No other book presents in the same space with the same clearness and exactness so large a range of mechanical principles.” Physical Review.
“Marked ability has been shewn in the development of the subject of Statics in the present volume.... The collected examples for students’ exercises are excellent.” Glasgow Herald.
584General Editors: F. H. Neville, M.A., F.R.S. and W. C. D. Whetham, M.A., F.R.S.
Mechanics. By John Cox, M.A., F.R.S.C., Macdonald Professor of Experimental Physics in McGill University, Montreal. Demy 8vo. pp. xiv + 332. 9s. Net.
Athenæum.—“It may reasonably be hoped that this endeavour to bridge over the gulf which has hitherto separated theory from practice in respect of the principles of mechanics, by showing their intimate connexion, and to present the subject in a more living and attractive form, by drawing attention to the gradual stages and methods by which the early investigators discovered the laws which govern the science, will meet with the success which it deserves.”
The Study of Chemical Composition. An Account of its Method and Historical Development, with illustrative quotations. By Ida Freund, Staff Lecturer and Associate of Newnham College. Demy 8vo. xvi + 650 pp. 18s. Net.
Saturday Review.—“Written from a broad, philosophical standpoint, we know of no book more suited for the student of chemistry who has attained a sound general knowledge of the science, and is now ready to appreciate a critical discussion of the methods by which the results he has learnt have been built up, thereby fitting himself for the real world of investigation on his own account.”
A Treatise on the Theory of Alternating Currents. By Alexander Russell, M.A., M.I.E.E.
Scotsman.—“The volume is not only rich in its own substantive teaching, but well supplied with references to the more remote authorities upon its subject. It opens an important and valuable contribution to the theoretical literature of electrical engineering.”
Radio-activity. By E. Rutherford, D.Sc., F.R.S., F.R.S.C., Macdonald Professor of Physics, McGill University, Montreal. Demy 8vo. pp. x + 400. 10s. 6d. Net.
Athenæum.—“English students have had to wait till now for any connected and detailed account of this new branch of physics from the pen of one who has a first hand knowledge of it.”
Nature.—“The arrangement of the matter and its treatment are throughout admirable.”
The Theory of Experimental Electricity. By W. C. D. Whetham, M.A., F.R.S., Fellow of Trinity College. Demy 8vo. 8s. Net.
1. Niewenglowski, C. R. 122, p. 385, 1896.
2. Becquerel, C. R. 122, p. 559, 1896.
3. Troost, C. R. 122, p. 564, 1896.
4. Arnold, Annal. d. Phys. 61, p. 316, 1897.
5. Le Bon, C. R. 122, pp. 188, 233, 386, 462, 1896.
6. Becquerel, C. R. 122, pp. 420, 501, 559, 689, 762, 1086, 1896.
7. Mme Curie, Thèse présentée à la Faculté des Sciences de Paris, 1903.
8. Nature, 56, 1897; Phil. Mag. 43, p. 418, 1897; 45, p. 277, 1898.
9. Rutherford, Phil. Mag. Jan. 1899.
10. Ibid.
11. Le Bon, C. R. 130, p. 891, 1900.
12. Lenard, Annal. d. Phys. 1, p. 498; 3, p. 298, 1900.
13. Schmidt, Annal. d. Phys. 65, p. 141, 1898.
14. Mme Curie, C. R. 126, p. 1101, 1898.
15. Owens, Phil. Mag. Oct. 1899.
16. Rutherford, Phil. Mag. Jan. 1900.
17. M. and Mme Curie and G. Bemont, C. R. 127, p. 1215, 1898.
18. Giesel, Phys. Zeit. 3, No. 24, p. 578, 1902.
19. Giesel, Annal. d. Phys. 69, p. 91, 1890. Ber. d. D. Chem. Ges. p. 3608, 1902.
20. Demarçay, C. R. 127, p. 1218, 1898; 129, p. 716, 1899; 131, p. 258, 1900.
21. Runge, Astrophys. Journal, p. 1, 1900. Annal. d. Phys. No. 10, p. 407, 1903.
22. Exner and Haschek, Wien. Ber. July 4, 1901.
23. Crookes, Proc. Roy. Soc. 72, p. 295, 1904.
24. Runge and Precht, Annal. d. Phys. XIV. 2, p. 418, 1904.
25. Runge and Precht, Phil. Mag. April, 1903.
26. Watts, Phil. Mag. July, 1903; August, 1904.
27. Runge, Phil. Mag. December, 1903.
28. Debierne, C. R. 129, p. 593, 1899; 130, p. 206, 1900.
29. Giesel, Ber. d. D. Chem. Ges. p. 3608, 1902; p. 342, 1903.
30. Debierne, C. R. 139, p. 538, 1904. Miss Brooks, Phil. Mag. Sept. 1904. Giesel, Phys. Zeit. 5, p. 822, 1904. Jahrbuch. d. Radioaktivität, no. 4, p. 345, 1904.
31. Giesel, Ber. d. D. Chem. Ges. 37, p. 1696, 1904; Hartmann, Phys. Zeit. 5, No. 18, p. 570, 1904.
32. Mme Curie, C. R. 127, p. 175, 1898.
33. Mme Curie, Thèse, Paris, 1903.
34. Crookes, Proc. Roy. Soc. May, 1900.
35. Berndt, Phys. Zeit. 2, p. 180, 1900.
36. Marckwald, Phys. Zeit. 4, No. 1 b, p. 51.
37. Marckwald, Ber. d. D. Chem. Ges. p. 2662, No. 12, 1903.
38. Elster and Geitel, Annal. d. Phys. 69, p. 83, 1899.
39. Giesel, Ber. d. D. Chem. Ges. p. 3775, 1901.
40. Hofmann and Strauss, Ber. d. D. Chem. Ges. p. 3035, 1901.
41. Hofmann, Gonder and Wölfl, Annal. d. Phys. No. 13, p. 615, 1904.
42. Hofmann and Zerban, Ber. d. D. Chem. Ges. No. 12, p. 3093, 1903.
43. Baskerville and Zerban, Amer. Chem. Soc. 26, p. 1642, 1904.
44. J. J. Thomson and Rutherford, Phil. Mag. Nov. 1896.
45. The word ion has now been generally adopted in the literature of the subject. In using this word, it is not assumed that the ions in gases are the same as the corresponding ions in the electrolysis of solutions.
46. A minute current is observed between the plates even if no radio-active matter be present. This has been found to be due mainly to a slight natural radio-activity of the matter composing them. (See chapter XIV.)
47. This nomenclature has arisen from the similarity of the shape of the current-voltage curves to the magnetization curves for iron. Since, on the ionization theory, the maximum current is a result of the removal of all the ions from the gas, before recombination occurs, the terms are not very suitable. They have however now come into general use and will be retained throughout this work.
48. J. J. Thomson, Phil. Mag. 47, p. 253, 1899; Conduction of Electricity through Gases, p. 73, 1903.
49. Rutherford, Phil. Mag. Jan. 1899.
50. Townsend, Phil. Mag. Feb. 1901.
51. Rutherford, Phil. Mag. Nov. 1897, p. 144, Jan. 1899.
52. Townsend, Phil. Trans. A, p. 157, 1899.
53. McClung, Phil. Mag. March, 1902.
54. Langevin, Thèse présentée à la Faculté des Sciences, p. 151, Paris, 1902.
55. Owens, Phil. Mag. Oct. 1899.
56. Rutherford, Phil. Mag. p. 429, Nov. 1897.
57. Zeleny, Phil. Trans. A, p. 193, 1901.
58. Langevin, C. R. 134, p. 646, 1902.
59. Zeleny, Phil. Mag. July, 1898.
60. Rutherford, Phil. Mag. Feb. 1899.
61. Zeleny, Phil. Trans. 195, p. 193, 1900.
62. Langevin, C. R. 134, p. 646, 1902, and Thesis, p. 191, 1902.
63. Rutherford, Proc. Camb. Phil. Soc. 9, p. 410, 1898.
64. Langevin, Thesis, p. 190, 1902.
65. Helmholtz and Richarz, Annal. d. Phys. 40, p. 161, 1890.
66. Wilson, Phil. Trans. p. 265, 1897; p. 403, 1899; p. 289, 1900.
67. Thomson, Phil. Mag. p. 528, Dec. 1898.
68. Wilson, Phil. Trans. A, 193, p. 289, 1899.
69. Thomson, Phil. Mag. p. 528, Dec. 1898, and March, 1903. Conduction of Electricity through Gases, Camb. Univ. Press, 1903, p. 121.
70. Wilson, Phil. Mag. April, 1903.
71. Townsend, Phil. Trans. A, p. 129, 1899.
72. Townsend, loc. cit. p. 139.
73. Some difference of opinion has been expressed as to the value of V required to produce ions at each collision. Townsend considers it to be about 20 volts; Langevin 60 volts and Stark about 50 volts.
74. Rutherford, Phil. Mag. Jan. 1899.
75. Rutherford, Phil. Mag. Jan. 1899.
76. Strutt, Phil. Trans. A, p. 507, 1901 and Proc. Roy. Soc. p. 208, 1903.
77. McClung, Phil. Mag. Sept. 1904.
78. Eve, Phil. Mag. Dec. 1904.
79. Rutherford, Phil. Mag. p. 137, Jan. 1899.
80. Child, Phys. Rev. Vol. 12, 1901.
81. Rutherford, Phil. Mag. p. 210, August, 1901; Phys. Rev. Vol. 13, 1901.
82. Rutherford, Phil. Mag. Aug. 1901.
83. A simple and excellent account of the effects produced by the motion of a charged ion and also of the electronic theory of matter was given by Sir Oliver Lodge in 1903 in a paper entitled “Electrons” (Proceedings of the Institution of Electrical Engineers, Part 159, Vol. 32, 1903). See also J. J. Thomson’s Electricity and Matter (Scribner, New York, 1904).
84. J. J. Thomson, Phil. Mag. April, 1887.
85. Heaviside, Collected Papers, Vol. II. p. 514.
86. Searle, Phil. Mag. Oct. 1897.
87. Abraham, Phys. Zeit. 4, No. 1 b, p. 57, 1902.
88. A full account of the path described by a moving ion under various conditions is given by J. J. Thomson, Conduction of Electricity in Gases (Camb. Univ. Press, 1903), pp. 79–90.
89. J. J. Thomson, Phil. Mag. p. 293, 1897.
90. Lenard, Annal. d. Phys. 64, p. 279, 1898.
91. Kaufmann, Annal. d. Phys. 61, p. 544; 62, p. 596, 1897; 65, p. 431, 1898.
92. Simon, Annal. d. Phys. 69, p. 589, 1899.
93. A complete discussion of the various methods employed to measure the velocity and mass of electrons and also of the theory on which they are based will be found in J. J. Thomson’s Conduction of Electricity through Gases.
94. Goldstein, Berlin Sitzber. 39, p. 691, 1896; Annal. d. Phys. 64, p. 45, 1898.
95. Wien, Annal. d. Phys. 65, p. 440, 1898.
96. Larmor, Phil. Mag. 44, p. 593, 1897.
97. J. J. Thomson, Phil. Mag. Feb. 1897.
98. Barkla, Phil. Mag. June, 1903.
99. Soddy, Trans. Chem. Soc. Vol. 81, p. 860, 1902.
100. Wilson, Proc. Roy. Soc. Vol. 68, p. 152, 1901.
101. If the apparatus is required to be air-tight, the gold-leaf system can be charged by means of a piece of magnetized steel wire, which is made to touch the rod R by the approach of a magnet.
102. It is sometimes observed that the motion of the gold-leaf, immediately after charging, is irregular. In many cases, this can be traced to air currents set up in the electroscope in consequence of unsymmetrical heating by the source of light used for illumination.
103. Wilson, Proc. Camb. Phil. Soc. Vol. 12, Part II. 1903.
104. Walker, Phil. Mag. Aug. 1903.
105. Strutt, Phil. Trans. A, p. 507, 1901.
106. Dolezalek, Instrumentenkunde, p. 345, Dec. 1901.
107. It is very desirable that care should be taken not to release large quantities of the radium emanation inside a laboratory. This emanation has a slow rate of decay and is carried by currents of air throughout the whole building and finally leaves behind an active deposit of very slow rate of change (see chapter XI.). Eve (Nature, March 16, 1905) has drawn attention to the difficulty of making refined radio-active measurements under such conditions.
108. J. J. Thomson, Phil. Mag. 46, p. 537, 1898.
109. Bronson, Amer. Journ. Science, Feb. 1905.
110. J. and P. Curie, C. R. 91, pp. 38 and 294, 1880. See also Friedel and J. Curie, C. R. 96, pp. 1262 and 1389, 1883, and Lord Kelvin, Phil. Mag. 36, pp. 331, 342, 384, 414, 453, 1893.
111. In an examination of uranium the writer (Phil. Mag. p. 116, Jan. 1899) found that the rays from uranium consist of two kinds, differing greatly in penetrating power, which were called the α and β rays. Later, it was found that similar types of rays were emitted by thorium and radium. On the discovery that very penetrating rays were given out by uranium and thorium as well as by radium, the term γ was applied to them by the writer. The word “ray” has been retained in this work, although it is now settled that the α and β rays consist of particles projected with great velocity. The term is thus used in the same sense as by Newton, who applied it in the Principia to the stream of corpuscles which he believed to be responsible for the phenomenon of light. In some recent papers, the α and β rays have been called the α and β “emanations.” This nomenclature cannot fail to lead to confusion, since the term “radio-active emanation” has already been generally adopted in radio-activity as applying to the material substance which gradually diffuses from thorium and radium compounds, and itself emits rays.
112. This method of illustration is due to Mme Curie, Thèse présentée à la Faculté des Sciences de Paris, 1903.
113. Giesel, Annal. d. Phys. 69, p. 834, 1899.
114. Meyer and Schweidler, Phys. Zeit. 1, pp. 90, 113, 1899.
115. Becquerel, C. R. 129, pp. 997, 1205, 1899.
116. Curie, C. R. 130, p. 73, 1900.
117. Rutherford, Phil. Mag. January, 1899.
118. Rutherford and Grier, Phil. Mag. September, 1902.
119. Becquerel, C. R. 130, pp. 206, 372, 810, 979. 1900.
120. M. and Mme Curie, C. R. 130, p. 647, 1900.
121. The activity of the radium preparation was not stated in the paper.
122. Dorn, Phys. Zeit. 4, No. 18, p. 507, 1903.
123. Strutt, Phil. Mag. Nov. 1903.
124. Wien, Phys. Zeit. 4, No. 23, p. 624, 1903.
125. Dorn, C. R. 130, p. 1129, 1900.
126. Becquerel, C. R. 130, p. 809, 1900.
127. Kaufmann, Phys. Zeit. 4, No. 1 b, p. 54, 1902.
128. Abraham, Phys. Zeit. 4, No. 1 b, p. 57, 1902.
129. Kaufmann, Nachrichten d. Ges. d. Wiss. zu Gött., Nov. 8, 1901.
130. Simon, Annal. d. Phys. p. 589, 1899.
131. Kaufmann, Phys. Zeit. 4, No. 1 b, p. 54, 1902.
132. Paschen, Annal. d. Phys. 14, p. 389, 1904.
133. Meyer and Schweidler, Phys. Zeit. pp. 90, 113, 209, 1900.
134. Lenard, Annal. d. Phys. 56, p. 275, 1895.
135. Strutt, Nature, p. 539, 1900.
136. Seitz, Phys. Zeit. 5, No. 14, p. 395, 1904.
137. It is presumed that the results were corrected, if necessary, for the discharging action due to the ionized gas, although no direct mention of this is made in the paper by Seitz.
138. Strutt, Phil. Trans. A, p. 507, 1901.
139. Crookes, Proc. Roy. Soc. 1902. Chem. News, 85, p. 109, 1902.
140. Mme Curie, C. R. 130, p. 76, 1900.
141. Rutherford, Phil. Mag. Feb. 1903. Phys. Zeit. 4, p. 235, 1902.
142. Becquerel, C. R. 136, p. 199, 1903.
143. Becquerel, C. R. 136, p. 431, 1903.
144. Des Coudres, Phys. Zeit. 4, No. 17, p. 483, 1903.
145. Becquerel, C. R. 136, p. 1517, 1903.
146. Bragg, Phil. Mag. Dec. 1904; Bragg and Kleeman, Phil. Mag. Dec. 1904.
147. Further experimental results bearing on this important question are given in an Appendix to this book.
148. Bakerian Lecture, Phil. Trans. A, p. 169, 1904.
149. Strutt, Phil. Mag. Aug. 1904.
150. J. J. Thomson, Proc. Camb. Phil. Soc. 13, Pt. I. p. 39, 1905. Nature, Dec. 15, 1904.
151. Rutherford, Nature, March 2, 1905. J. J. Thomson, Nature, March 9, 1905.
152. Crookes, Proc. Roy. Soc. 81, p. 405, 1903.
153. Elster and Geitel, Phys. Zeit. No. 15, p. 437, 1903.
154. Glew, Arch. Röntgen Ray, June 1904.
155. Becquerel, C. R. 137, Oct. 27, 1903.
156. Tommasina, C. R. 137, Nov. 9, 1903.
157. An interesting side-light is thrown on this question by the experiments described in Appendix A of this book.
158. Rutherford and Miss Brooks, Phil. Mag. July 1902.
159. In order to obtain a thin layer, the compound to be tested is ground to a fine powder and then sifted through a fine gauge uniformly over the area, so that the plate is only partially covered.
160. Rutherford, Phil. Mag. Jan. 1899.
161. Owens, Phil. Mag. Oct. 1899.
162. Rutherford and Miss Brooks, Phil. Mag. July, 1900.
163. Since the ionization at any point above the plate is the resultant effect of the α particles coming from all points of the large radio-active layer, λ is not the same as the coefficient of absorption of the rays from a point source. It will however be proportional to it. For this reason λ is called the “absorption constant.”
164. Townsend, Phil. Mag. Feb. 1901.
165. Durack, Phil. Mag. July 1902, May 1903.
166. Bragg and Bragg and Kleeman, Phil. Mag. Dec. 1904.
167. Villard, C. R. 130, pp. 1010, 1178, 1900.
168. Becquerel, C. R. 130, p. 1154, 1900.
169. Rutherford, Phys. Zeit. 3, p. 517, 1902.
170. McClelland, Phil. Mag. July 1904.
171. Paschen, Phys. Zeit. 5, No. 18, p. 563, 1904.
172. A. S. Eve, Phil. Mag. Nov. 1904.
173. Paschen, Annal. d. Physik, 14, p. 114, 1904; 14, 2, p. 389, 1904. Phys. Zeit. 5, No. 18, p. 563, 1904.
174. Paschen, Phys. Zeit. 5, No. 18, p. 563, 1904.
175. Rutherford and Barnes, Phil. Mag. May 1905. Nature, p. 151, Dec. 15, 1904.
176. Barkla, Nature, March 17, 1904.
177. Becquerel, C.R. 132, pp. 371, 734, 1286. 1901.
178. Mme Curie, Thèse présentée à la Faculté des Sciences, Paris 1903, p. 85.
179. A. S. Eve, Phil. Mag. Dec. 1904.
180. In a recent paper (Phil. Mag. Feb. 1905), McClelland has, in the main, confirmed the experimental results obtained by Eve. An electrometer was used instead of an electroscope. He finds, in addition, that the amount of secondary radiation depends on the angle of incidence of the primary rays, and is greatest for an angle of 45°. In a letter to Nature (Feb. 23, p. 390, 1905), he states that more recent experiments have shown that the amount of secondary radiation from different substances is a function of their atomic weights rather than of their densities. In every case examined, the amount of secondary radiation increases with the atomic weight, but is not proportional to it.
181. Rutherford and McClung, Phil. Trans. A. p. 25, 1901.
182. Meyer and Schweidler, Wien Ber. 113, July, 1904.
183. Rutherford and Grier, Phil. Mag. Sept. 1902.
184. Becquerel, C. R. 129, p. 912, 1899.
185. Bary, C. R. 130, p. 776, 1900.
186. Kunz and Baskerville, Amer. Journ. Science XVI. p. 335, 1903.
187. See Nature, p. 523, March 31, 1904.
188. Crookes, Proc. Roy. Soc. 74, p. 47, 1904.
189. Kunz and Baskerville, Science XVIII, p. 769, Dec. 18, 1903.
190. Beilby in a recent communication to the Royal Society (Feb. 9 and 23, 1905) has examined in some detail the production of phosphorescence by the β and γ rays of radium and has put forward a theory to account for the different actions observed.
191. Huggins, Proc. Roy. Soc. 72, pp. 196 and 409, 1903.
192. The spark spectrum of the radium bromide showed the H and K lines of calcium and also faintly some of the strong lines of barium. The characteristic lines of radium of wave-lengths 3814·59, 3649·7, 4340·6 and 2708·6, as shown by Demarçay and others are clearly shown in the figure. The strong line of wave-length about 2814 is due to radium.
193. Giesel, Ber. d. D. Chem. Ges. 37, p. 1696, 1904.
194. Hartmann, Phys. Zeit. 5, No. 18, p. 570, 1904.
195. In a recent paper, Giesel (Ber. d. D. Chem. Ges. No. 3, p. 775, 1905) has shown that the bright lines are due to didymium, which is present as an impurity. Exposure of didymium to the radium rays also causes the appearance of the lines.
196. Wiedemann and Schmidt, Wied. Annal. 59, p. 604, 1895.
197. Wiedemann, Phys. Zeit. 2, p. 269, 1901.
198. Elster and Geitel, Annal. d. Phys. 69, p. 673, 1899.
199. Willons and Peck (Phil. Mag. March, 1905) found that under some conditions, especially for long sparks, the rays of radium hindered the passage of the spark.
200. Hemptinne, C. R. 133, p. 934, 1901.
201. Himstedt, Phys. Zeit. p. 476, 1900.
202. Henning, Annal. d. Phys. p. 562, 1902.
203. Kohlrausch and Henning, Verh. Deutsch. Phys. Ges. 6, p. 144, 1904.
204. Kohlrausch, Verh. Deutsch. Phys. Ges. 5, p. 261, 1904.
205. P. Curie, C. R. 134, p. 420, 1902.
206. Becquerel, C. R. 136, p. 1173, 1903.
207. Becquerel, C. R. 133, p. 199, 1901.
208. P. Curie, Société de Physique, March 2, 1900.
209. Joly, Phil. Mag. March, 1904.
210. S. and P. Curie, C. R. 129, p. 823, 1899.
211. Giesel, Verhandlg. d. D. Phys. Ges. Jan. 5, 1900.
212. Salomonsen and Dreyer, C. R. 139, p. 533, 1904.
213. Elster and Geitel, Phys. Zeit. p. 113, No. 3, 1902.
214. Becquerel, C. R. 133, p. 709, 1901.
215. Hardy and Miss Wilcock, Proc. Roy. Soc. 72, p. 200, 1903.
216. Hardy, Proc. Physiolog. Soc. May 16, 1903.
217. Whetham, Phil. Mag. Nov. 1899; Theory of Solution, Camb. 1902, p. 396.
218. Curie and Debierne, C. R. 132, p. 768, 1901.
219. Giesel, Ber. D. d. Chem. Ges. 35, p. 3605, 1902.
220. Ramsay and Soddy, Proc. Roy. Soc. 72, p. 204, 1903.
221. Danysz, C. R. 136, p. 461, 1903.
222. Aschkinass and Caspari, Arch. d. Ges. Physiologie, 86, p. 603, 1901.
223. Himstedt and Nagel, Drude’s Annal. 4, p. 537, 1901.
224. Hardy and Anderson, Proc. Roy. Soc. 72, p. 393, 1903.
225. Crookes, Proc. Roy. Soc. 66, p. 409, 1900.
226. Becquerel, C. R. 131, p. 137, 1900; 133, p. 977, 1901.
227. Rutherford and Soddy, Phil. Mag. Sept. and Nov. 1902. Trans. Chem. Soc. 81, pp. 321 and 837, 1902.
228. Rutherford and Soddy, Phil. Mag. Sept. 1902.
229. The general method of regarding the subject would be unchanged, even if it were proved that the radio-activity of thorium is not due to thorium at all but to a small constant amount of a radio-active impurity mixed with it.
230. Rutherford and Soddy, Phil. Mag. Sept. 1902.
231. Owens, Phil. Mag. p. 360, Oct. 1899.
232. Rutherford, Phil. Mag. p. 1, Jan. 1900.
233. Rossignol and Gimingham, Phil. Mag. July, 1904.
234. Bronson, Amer. Journ. Science, Feb. 1905.
235. Phil. Mag. April, 1904.
236. Dorn, Abh. der. Naturforsch. Ges. für Halle-a-S., 1900.
237. P. Curie, C. R. 135, p. 857, 1902.
238. Rutherford and Soddy, Phil. Mag. April, 1903.
239. P. Curie, C. R. 136, p. 223, 1903.
240. Debierne, C. R. 136, p. 146, 1903.
241. Giesel, Ber. D. deutsch. Chem. Ges. p. 3608, 1902.
242. Curie and Debierne, C. R. 132, pp. 548 and 768, 1901.
243. Curie and Debierne, C. R. 133, p. 931, 1901.
244. Rutherford and Soddy, Trans. Chem. Soc. p. 321, 1902. Phil. Mag. Sept. 1902.
245. Rutherford, Phys. Zeit. 2, p. 429, 1901.
246. Rutherford and Soddy, Phil. Mag. Nov. 1902.
247. Rutherford and Soddy, Phil. Mag. April, 1903.
248. Rutherford and Soddy, Phil. Mag. Nov. 1902.
249. Rutherford and Soddy, Phil. Mag. April, 1903.
250. Curie and Debierne, C. R. 133, p. 931, 1901.
251. Rutherford and Soddy, Phil. Mag. Nov. 1902.
252. Ramsay and Soddy, Proc. Roy. Soc. 72, p. 204, 1903.
253. Rutherford and Miss Brooks, Trans. Roy. Soc. Canada 1901, Chem. News 1902.
254. Loschmidt, Sitzungsber. d. Wien. Akad. 61, II. p. 367, 1871.
255. See Stefan, Sitzungsber. d. Wien. Akad. 63, II. p. 82, 1871.
256. P. Curie and Danne, C. R. 136, p. 1314, 1903.
257. Bumstead and Wheeler, Amer. Jour. Science, Feb. 1904.
258. Makower, Phil. Mag. Jan. 1905.
259. Wallstabe, Phys. Zeit. 4, p. 721, 1903.
260. Stefan, Wien. Ber. 2, p. 371, 1878.
261. Rutherford and Soddy, Phil. Mag. Nov. 1902.
262. Phil. Mag. May, 1903.
263. P. Curie, Société de Physique, 1903.
264. Rutherford and Soddy, Phil. Mag. May, 1903.
265. Nature, Aug. 20, 1903.
266. Proc. Roy. Soc. 73, No. 494, p. 346, 1904.
267. Proc. Roy. Soc. 73, No. 495, p. 470, 1904.
268. Pickering, Astrophys. Journ. Vol. 14, p. 368, 1901.
269. M. and Mme. Curie, C. R. 129, p. 714, 1899.
270. Rutherford, Phil. Mag. Jan. and Feb. 1900.
271. As regards date of publication, the priority of the discovery of “excited activity” belongs to M. and Mme. Curie. A short paper on this subject, entitled “Sur la radioactivité provoquée par les rayons de Becquerel,” was communicated by them to the Comptes Rendus, Nov. 6, 1899. A short note was added to the paper by Becquerel in which the phenomena of excited activity were ascribed to a type of phosphorescence. On my part, I had simultaneously discovered the emission of an emanation from thorium compounds and the excited activity produced by it, in July, 1899. I, however, delayed publication in order to work out in some detail the properties of the emanation and of the excited activity and the connection between them. The results were published in two papers in the Philosophical Magazine (Jan. and Feb. 1900) entitled “A radio-active substance emitted from thorium compounds,” and “Radio-activity produced in substances by the action of thorium compounds.”
272. Rutherford, Phil. Mag. Feb. 1900.
273. Rutherford, Phys. Zeit. 3, No. 12, p. 254, 1902. Phil. Mag. Jan. 1903.
274. Miss Brooks, Phil. Mag. Sept. 1904.
275. Rutherford and Miss Brooks, Phil. Mag. July, 1902.
276. Curie and Danne, C. R. 136, p. 364, 1903.
277. Mme Curie, Thèse, Paris, 1903, p. 116.
278. Debierne, C. R. 138, p. 411, 1904.
279. Giesel, Ber. d. D. Chem. Ges. No. 3, p. 775, 1905.
280. Miss Brooks, Phil. Mag. Sept. 1904.
281. Rutherford, Phys. Zeit. 3, No. 12, p. 254, 1902.
282. F. von Lerch, Annal. d. Phys. 12, p. 745, 1903.
283. Pegram, Phys. Review, p. 424, Dec. 1903.
284. Miss Gates, Phys. Review, p. 300, 1903.
285. A more complete examination of the effect of temperature on the excited activity of thorium has been made by Miss Slater (section 207).
286. Rutherford, Phil. Mag. Feb. 1900.
287. Henning, Annal. d. Phys. 7, p. 562, 1902.
288. Rutherford, Phil. Mag. Feb. 1900.
289. Curie and Debierne, C. R. 132, p. 768, 1901.
290. Fehrle, Phys. Zeit. 3, No. 7, p. 130, 1902.
291. Rutherford, Phil. Mag. Jan. 1903.
292. Giesel, Ber. d. D. Chem. Ges. 36, p. 342, 1903.
293. Debierne, C. R. 136, pp. 446 and 671, 1903; 138, p. 411, 1904.
294. Ramsay, Proc. Roy. Soc. p. 470, June, 1904; C. R. 138, June 6, 1904.
295. Phil. Mag. February, 1904.
296. Soddy, Trans. Chem. Soc. 81, p. 460, 1902.
297. Rutherford and Grier, Phil. Mag. Sept. 1902.
298. Becquerel, C. R. 131, p. 137, 1900.
299. Meyer and Schweidler, Wien Ber. Dec. 1, 1904.
300. Meyer and Schweidler, Wien Ber. 113, July, 1904.
301. Rutherford, Phil. Trans. A. 204, pp. 169–219, 1904.
302. Pegram, Phys. Rev. p. 424, December, 1903.
303. Miss Slater, Phil. Mag. 1905.
304. von Lerch, Ann. de d. Phys. November, 1903.
305. The ‘rayless change’ certainly does not give out α rays, and special experiments showed that no appreciable amount of β rays were present. On the other hand, the second change gives out all three types of rays.
306. Miss Brooks, Phil. Mag. Sept. 1904.
307. Rutherford and Soddy, Trans. Chem. Soc. 81, p. 837, 1902. Phil. Mag. Nov. 1902.
308. Miss Brooks, Phil. Mag. Sept. 1904.
309. Rutherford, Phil. Trans. A. p. 169, 1904.
310. Giesel, Ber. d. D. Chem. Ges. p. 775, 1905.
311. Godlewski, Nature, p. 294, Jan. 19, 1905.
312. Debierne, C. R. 138, p. 411, 1904.
313. Miss Brooks, Phil. Mag. Sept. 1904.
314. Rutherford and Soddy, Phil. Mag. April, 1903.
315. Rutherford, Phil. Trans. A. p. 169, 1904. Curie and Danne, C. R. p. 748, 1904.
316. P. Curie and Danne, Comptes Rendus, 138, p. 748, 1904.
317. Miss Gates, Phys. Rev. p. 300, 1903.
318. Miss Brooks, Nature, July 21, 1904.
319. Rutherford, Phil. Mag. Nov. 1904. Nature, p. 341, Feb. 9, 1905.
320. Rutherford, Nature, p. 341, Feb. 9, 1905.
321. Marckwald (Ber. d. D. Chem. Ges. p. 591, 1905) has recently found that the activity of his radio-tellurium falls to half value in 139 days.
322. Meyer and Schweidler, Wien Ber. Dec. 1, 1904.
323. Rutherford, Phil. Trans. A. p. 169, 1904.
324. Hofmann, Gonder and Wölfl, Annal. d. Phys. 15, p. 615, 1904.
325. Phil. Trans. A. p. 25, 1901.
326. P. Curie and Laborde, C. R. 136, p. 673, 1903.
327. Runge and Precht, Sitz. Ak. Wiss. Berlin, No. 38, 1903.
328. P. Curie, Société de Physique, 1903.
329. Rutherford and Barnes, Nature, Oct. 29, 1903. Phil. Mag. Feb. 1904.
330. Paschen, Phys. Zeit. Sept. 15, 1904.
331. Rutherford and Barnes, Nature, Dec. 18, 1904; Phil. Mag. May, 1905.
332. Pegram, Science, May 27, 1904.
333. Perrin, Revue Scientifique, April 13, 1901.
334. Becquerel, C. R. 133, p. 979, 1901.
335. Rutherford and McClung, Phil. Trans. A, p. 25, 1901.
336. Rutherford, Phil. Mag. Jan. and Feb. 1900.
337. P. Curie, C. R. 136, p. 223, 1903.
338. Rutherford, Phil. Mag. April, 1903.
339. M. and Mme Curie, C. R. 134, p. 85, 1902.
340. Rutherford and Soddy, Trans. Chem. Soc. 81, pp. 321, 837, 1902. Phil. Mag. Sept. and Nov. 1902.
341. Rutherford and Soddy, Phil. Mag. April, 1903.
342. Rutherford and Soddy, Phil. Mag. May, 1903.
343. Rutherford, Phys. Zeit. 4, p. 235, 1902. Phil. Mag. Feb. 1903.
344. Rutherford, Phil. Mag. May, 1903.
345. Curie and Laborde, C. R. 136, p. 673, 1903.
346. J. J. Thomson, Nature, p. 601, 1903.
347. Crookes, C. R. 128, p. 176, 1899.
348. F. Re, C. R. p. 136, p. 1393, 1903.
349. Richarz and Schenck, Berl. Ber. p. 1102, 1903. Schenck, Berl. Ber. p. 37, 1904.
350. Armstrong and Lowry, Proc. Roy. Soc. 1903. Chem. News, 88, p. 89, 1903.
351. Rutherford and Soddy, Phil. Mag. May, 1903.
352. Boltwood, Nature, May 25, p. 80, 1904. Phil. Mag. April, 1905.
353. McCoy, Ber. d. D. Chem. Ges. No. 11, p. 2641, 1904.
354. Strutt, Nature, March 17 and July 7, 1904. Proc. Roy. Soc. March 2, 1905.
355. Strutt, Proc. Roy. Soc. March 2, 1905.
356. Soddy, Nature, May 12, 1904; Jan. 19, 1905.
357. Whetham, Nature, May 5, 1904; Jan. 26, 1905.
358. Danne, C. R. Jan. 23, 1905.
359. J. J. Thomson, Nature, April 30, p. 601, 1903.
360. Voller, Phys. Zeit. 5, No. 24, p. 781, 1904.
361. Ramsay and Cooke, Nature, Aug. 11, 1904.
362. Eve, Nature, March 16, 1905.
363. J. J. Thomson, International Electrical Congress, St Louis, Sept. 1904.
364. Rutherford and Soddy, Phil. Mag. p. 582, 1902; pp. 453 and 579, 1903.
365. Ramsay and Soddy, Nature, July 16, p. 246, 1903. Proc. Roy. Soc. 72, p. 204, 1903; 73, p. 346, 1904.
366. Curie and Dewar, C. R. 138, p. 190, 1904. Chem. News, 89, p. 85, 1904.
367. Himstedt and Meyer, Ann. d. Phys. 15, p. 184, 1904.
368. Strutt, Proc. Roy. Soc. March 2, 1905.
369. Boltwood, Phil. Mag. April, 1905.
370. Moss, Trans. Roy. Soc. Dublin, 1904.
371. Travers, Nature, p. 248, Jan. 12, 1905.
372. Jaquerod, C. R. p. 789, 1904.
373. Ramsay and Travers, Zeitsch. Physik. Chem. 25, p. 568, 1898.
374. Ramsay, Nature, April 7, 1904.
375. Lodge, Nature, June 11, p. 129, 1903.
376. Larmor, Aether and Matter, p. 233.
377. J. J. Thomson, Phil. Mag. p. 681, Dec. 1903.
378. Lord Kelvin, Phil. Mag. Oct. 1904.
379. Thomson, Phil. Mag. March, 1904.
380. Rutherford and Soddy, Phil. Mag. May, 1903.
381. See Strutt and Joly, Nature, Oct. 15, 1903.
382. Strutt, Phil. Mag. June, 1903.
383. Elster and Geitel, Phys. Zeit. 4, No. 19, p. 522, 1903. Chem. News, July 17, p. 30, 1903.
384. Geitel, Phys. Zeit. 2, p. 116, 1900.
385. C. T. R. Wilson, Proc. Camb. Phil. Soc. 11, p. 32, 1900. Proc. Roy. Soc. 68, p. 151, 1901.
386. Elster and Geitel, Phys. Zeit. 2, p. 590, 1901.
387. Elster and Geitel, Phys. Zeit. 3, p. 76, 1901.
388. Rutherford and Allan, Phil. Mag. Dec. 1902.
389. Allan, Phil. Mag. Feb. 1904.
390. C. T. R. Wilson, Proc. Camb. Phil. Soc. 11, p. 428, 1902.
391. C. T. R. Wilson, Proc. Camb. Phil. Soc. 11, p. 428, 1902; 12, p. 17, 1903.
392. C. T. R. Wilson, Proc. Camb. Phil. Soc. 12, p. 85, 1903.
393. Allan, Phys. Rev. 16, p. 106, 1903.
394. McLennan, Phys. Rev. 16, p. 184, 1903.
395. Schmauss, Annal. d. Phys. 9, p. 224, 1902.
396. Elster and Geitel, Phys. Zeit. 3, p. 574, 1902.
397. Ebert and Ewers, Phys. Zeit. 4, p. 162, 1902.
398. Sarasin, Tommasina and Micheli, C. R. 139, p. 917, 1905.
399. J. J. Thomson, Phil. Mag. Sept. 1902.
400. Ebert, Sitz. Akad. d. Wiss. Munich, 33, p. 133, 1903.
401. J. J. Thomson, Phil. Mag. Sept. 1902.
402. Adams, Phil. Mag. Nov. 1903.
403. Bumstead and Wheeler, Amer. Journ. Science, 17, p. 97, Feb. 1904.
404. Bumstead, Amer. Journ. Science, 18, July, 1904.
405. Dadourian, Amer. Journ. Science, 19, Jan. 1905.
406. H. S. Allen and Lord Blythswood, Nature, 68, p. 343, 1903; 69, p. 247, 1904.
407. Strutt, Proc. Roy. Soc. 73, p. 191, 1904.
408. Himstedt, Ann. d. Phys. 13, p. 573, 1904.
409. Elster and Geitel, Phys. Zeit. 5, No. 12, p. 321, 1904.
410. Dorn, Abhandl. d. Natur. Ges. Halle, 25, p. 107, 1904.
411. Schenck, Thesis Univ. Halle, 1904.
412. Mache, Wien. Ber. 113, p. 1329, 1904.
413. Curie and Laborde, C. R. 138, p. 1150, 1904.
414. Blanc, Phil. Mag. Jan. 1905.
415. Boltwood, Amer. Journ. Science, 18, Nov. 1904.
416. Elster and Geitel, Phys. Zeit. 4, p. 522, 1903.
417. Elster and Geitel, Phys. Zeit. 5, No. 1, p. 11, 1903.
418. Vincenti and Levi Da Zara, Atti d. R. Instit. Veneto d. Scienze, 54, p. 95, 1905.
419. Burton, Phil. Mag. Oct. 1904.
420. Elster and Geitel, Phys. Zeit. 6, No. 3, p. 67, 1905.
421. Rutherford and Allan, Phil. Mag. Dec. 1902.
422. Elster and Geitel, Phys. Zeit. 4, p. 138, 1902; 4, p. 522, 1903.
423. Saake, Phys. Zeit. 4, p. 626, 1903.
424. Simpson, Proc. Roy. Soc. 73, p. 209, 1904.
425. McLennan, Phys. Rev. 16, p. 184, 1903, and Phil. Mag. 5, p. 419, 1903.
426. McLennan, Phys. Rev. No. 4, 1903.
427. Rutherford and Cooke, Americ. Phys. Soc. Dec. 1902.
428. Cooke, Phil. Mag. Oct. 1903.
429. Allan, Phil. Mag. Feb. 1904.
430. Ebert, Phys. Zeit. 2, p. 622, 1901. Zeitschr. f. Luftschiffahrt, 4, Oct. 1902.
431. Schuster, Proc. Manchester Phil. Soc. p. 488, No. 12, 1904.
432. Mache and Von Schweidler, Phys. Zeit. 6, No. 3, p. 71, 1905.
433. Langevin, C. R. 140, p. 232, 1905.
434. Schuster, British Assoc. 1903.
435. J. J. Thomson, Conduction of Electricity through Gases, p. 324, 1903.
436. Miss Gates, Phys. Rev. 17, p. 499, 1903.
437. Villard, Société de Physique, July, 1900.
438. Geitel, Phys. Zeit. 2, p. 116, 1900.
439. C. T. R. Wilson, Proc. Camb. Phil. Soc. 11, p. 52, 1900. Proc. Roy. Soc. 68, p. 152, 1901.
440. Rutherford and Allan, Phil. Mag. Dec. 1902.
441. Patterson, Phil. Mag. August, 1903.
442. Harms, Phys. Zeit. 4, No. 1, p. 11, 1902.
443. Cooke, Phil. Mag. Oct. 1903.
444. Wilson, Proc. Roy. Soc. 69, p. 277, 1901.
445. Jaffé, Phil. Mag. Oct. 1904.
446. Patterson, Phil. Mag. Aug. 1903.
447. Strutt, Phil. Mag. June, 1903. Nature, Feb. 19, 1903.
448. McLennan and Burton, Phys. Rev. No. 4, 1903. J. J. Thomson, Nature, Feb. 26, 1903.
449. Cooke, Phil. Mag. Aug. 6, 1903. Rutherford, Nature, April 2, 1903.
450. Eve, Nature, March 16, 1905.
451. See article in Le Radium, No. 3, p. 81, Sept. 15, 1904.
452. J. J. Thomson, Proc. Camb. Phil. Soc. 12, p. 391, 1904.
453. Wood, Phil. Mag. April, 1905.
454. Campbell, Nature, p. 511, March 31, 1904. Phil. Mag. April, 1905.
455. An apparent exception has been observed by Danne in the case of certain lead minerals which occur under peculiar conditions at d’Issy-l’Évêque, France. See p. 465.